

p53-mediated neurodegeneration in the absence of the nuclear protein Akirin2
- PMID: 35198879
- PMCID: PMC8844820
- DOI: 10.1016/j.isci.2022.103814
p53-mediated neurodegeneration in the absence of the nuclear protein Akirin2
Abstract
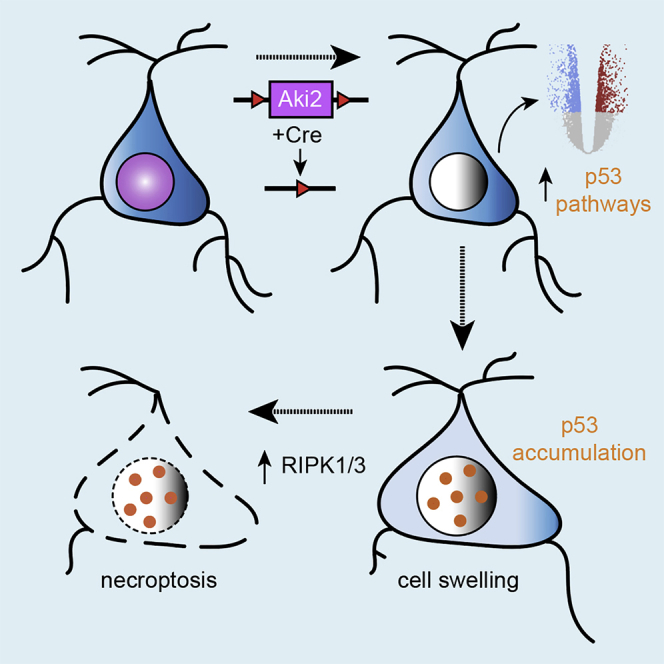
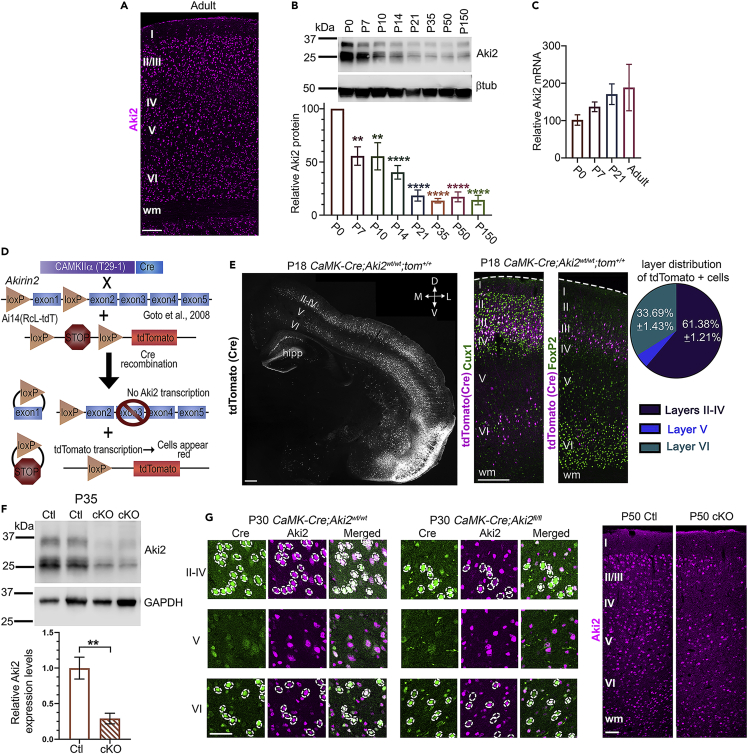
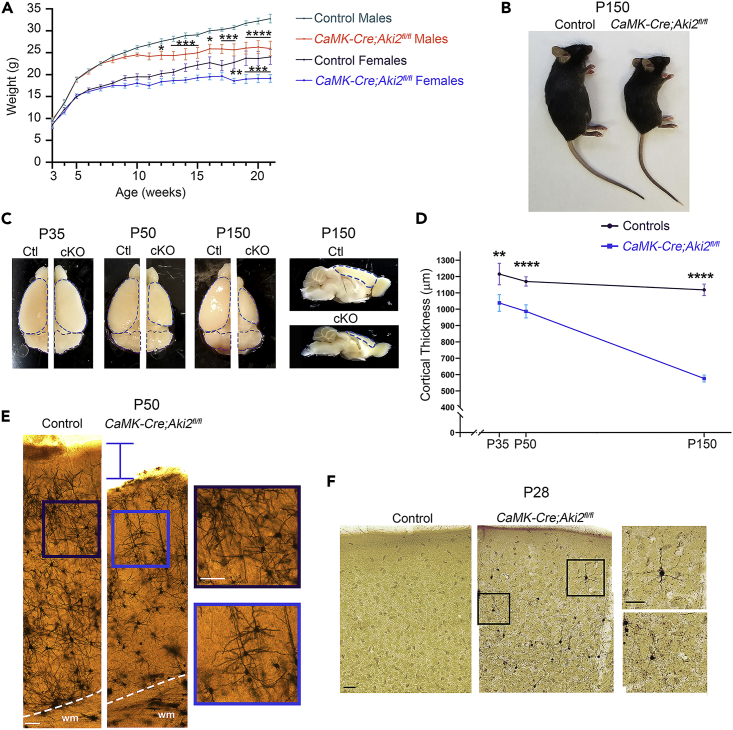
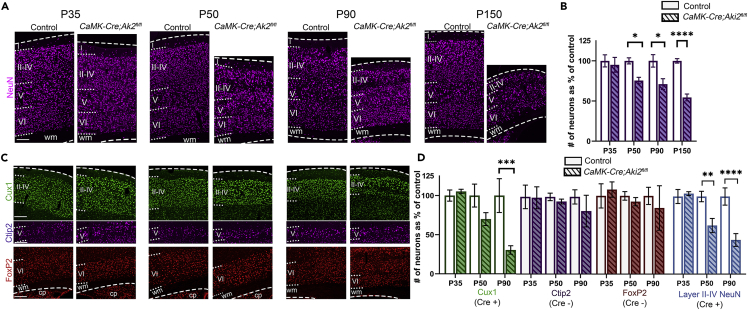
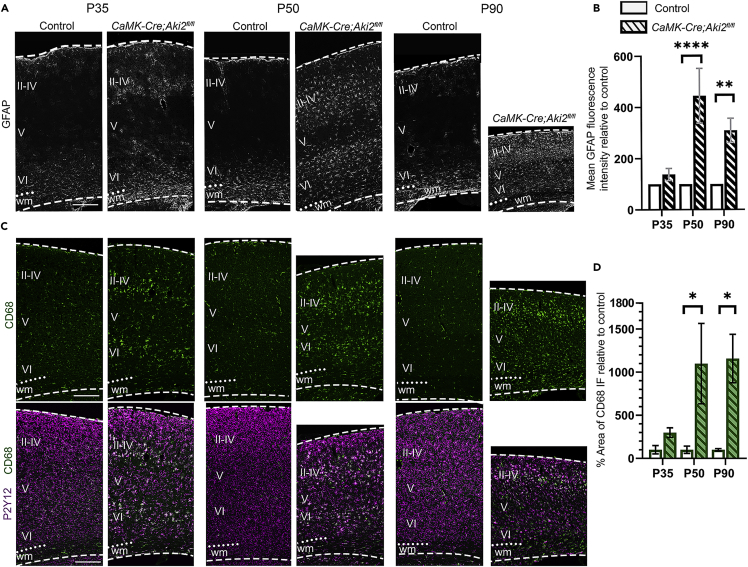
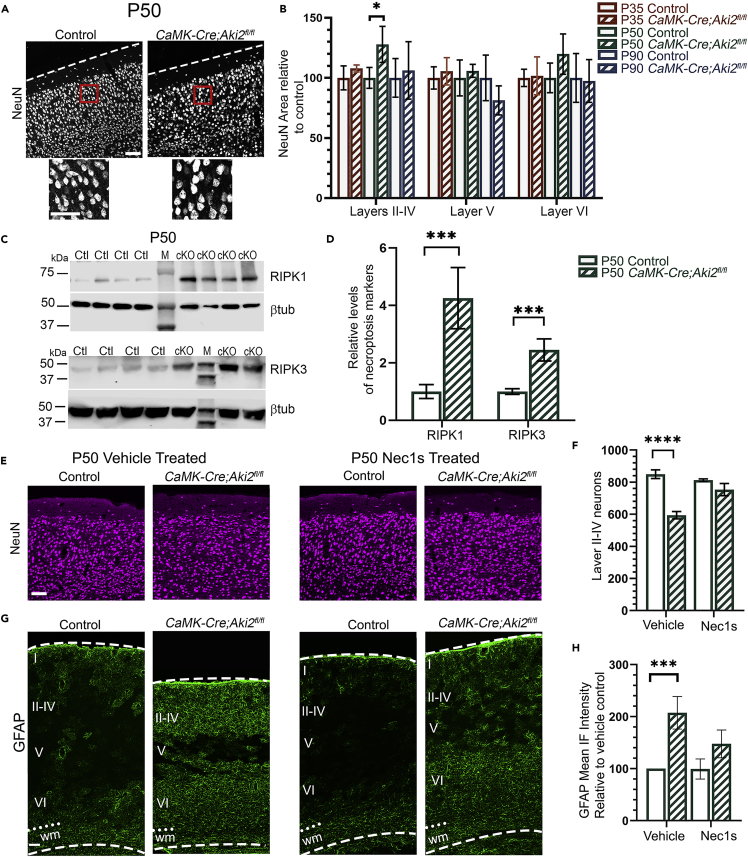
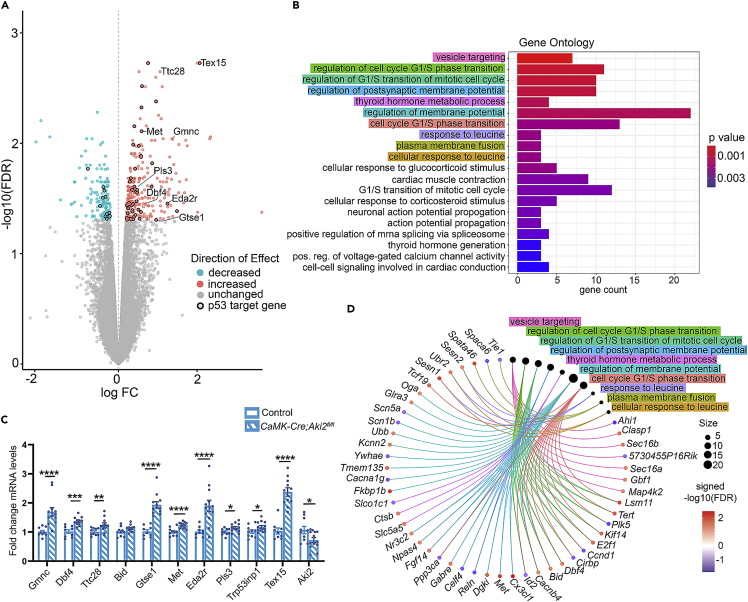
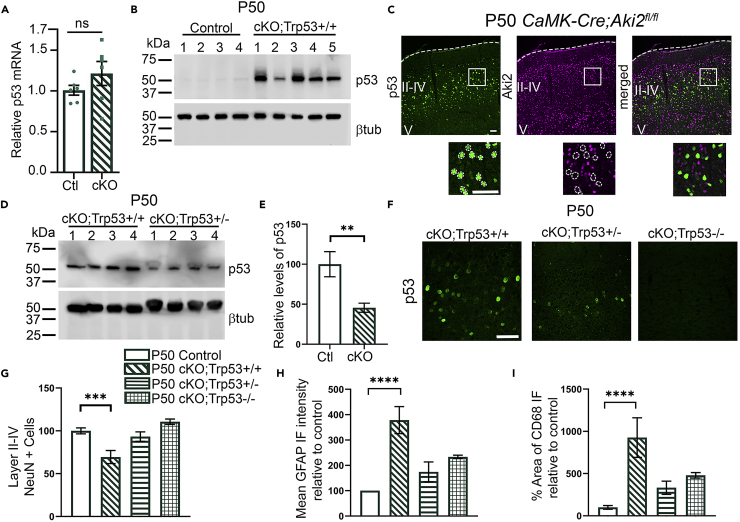
Proper gene regulation is critical for both neuronal development and maintenance as the brain matures. We previously demonstrated that Akirin2, an essential nuclear protein that interacts with transcription factors and chromatin remodeling complexes, is required for the embryonic formation of the cerebral cortex. Here we show that Akirin2 plays a mechanistically distinct role in maintaining healthy neurons during cortical maturation. Restricting Akirin2 loss to excitatory cortical neurons resulted in progressive neurodegeneration via necroptosis and severe cortical atrophy with age. Comparing transcriptomes from Akirin2-null postnatal neurons and cortical progenitors revealed that targets of the tumor suppressor p53, a regulator of both proliferation and cell death encoded by Trp53, were consistently upregulated. Reduction of Trp53 rescued neurodegeneration in Akirin2-null neurons. These data: (1) implicate Akirin2 as a critical neuronal maintenance protein, (2) identify p53 pathways as mediators of Akirin2 functions, and (3) suggest Akirin2 dysfunction may be relevant to neurodegenerative diseases.
Keywords: biological sciences; cell biology; molecular biology; neuroscience.
© 2022 The Author(s).
Conflict of interest statement
The authors declare no competing interests
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
