

DCHS1, Lix1L, and the Septin Cytoskeleton: Molecular and Developmental Etiology of Mitral Valve Prolapse
- PMID: 35200715
- PMCID: PMC8874669
- DOI: 10.3390/jcdd9020062
DCHS1, Lix1L, and the Septin Cytoskeleton: Molecular and Developmental Etiology of Mitral Valve Prolapse
Abstract
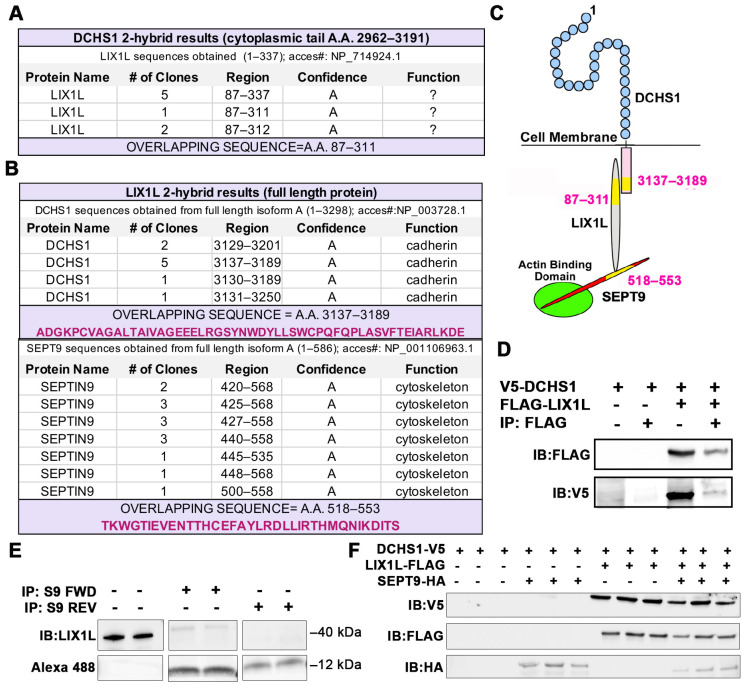
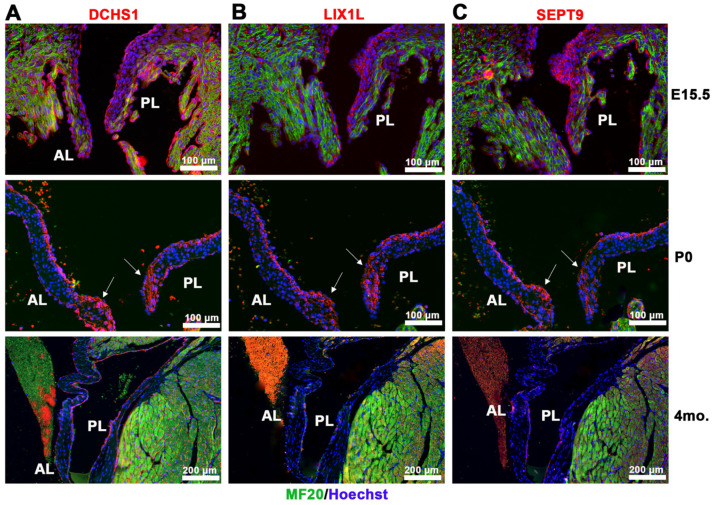
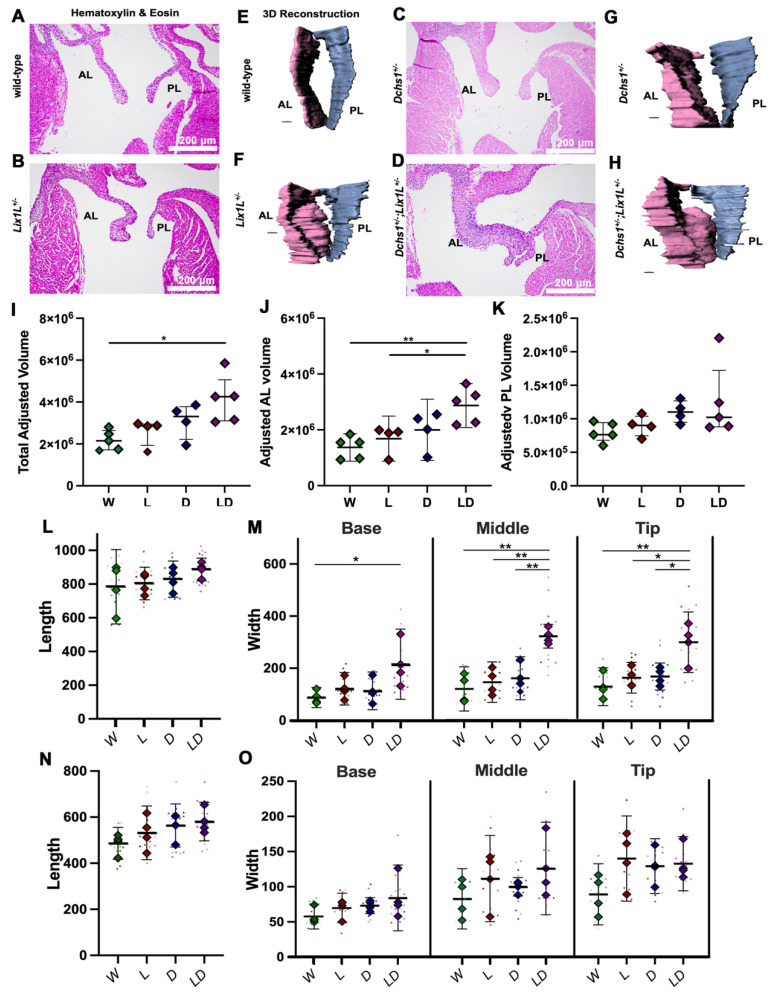
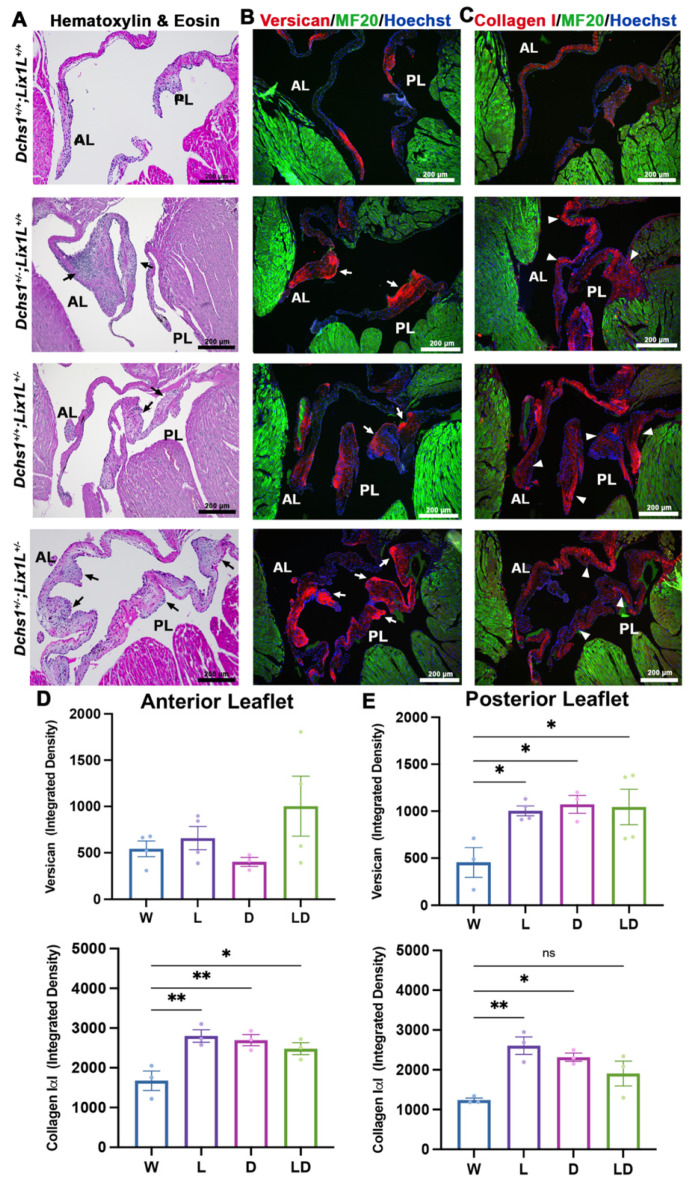
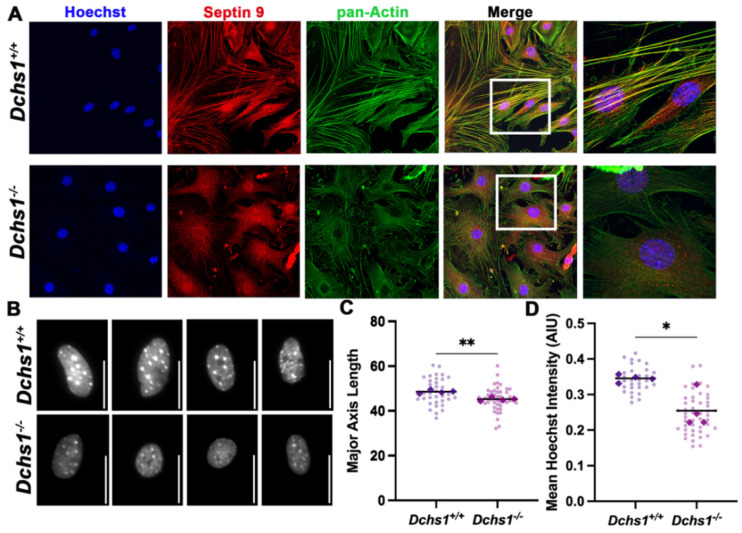
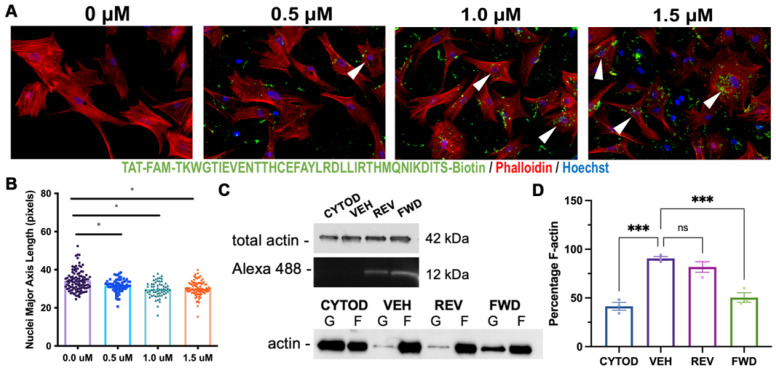
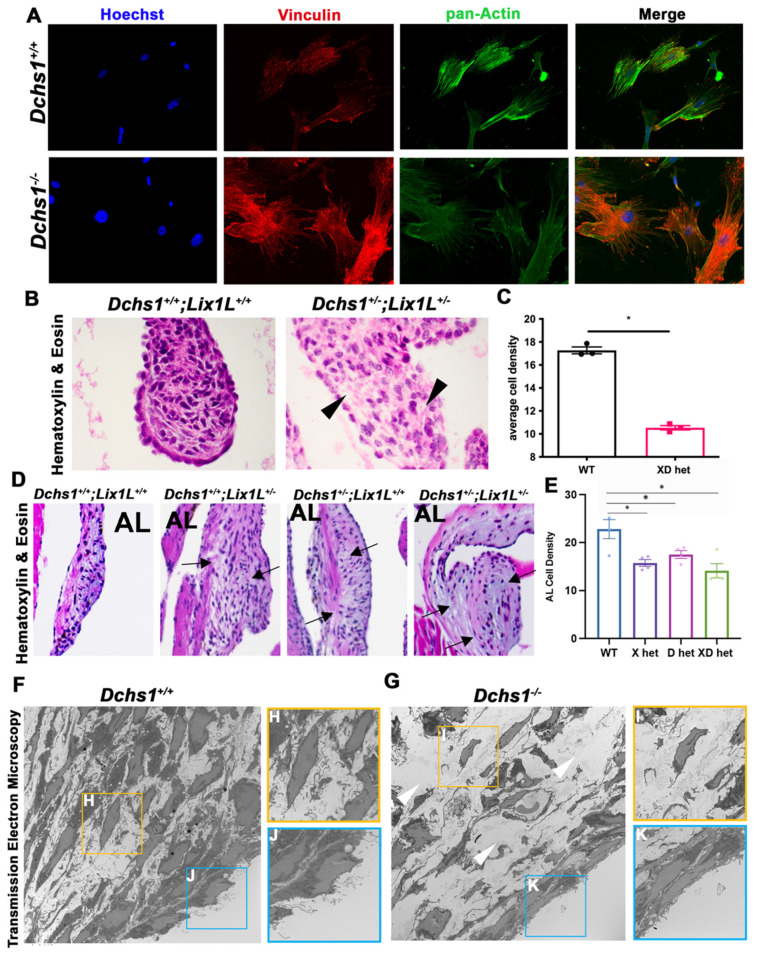
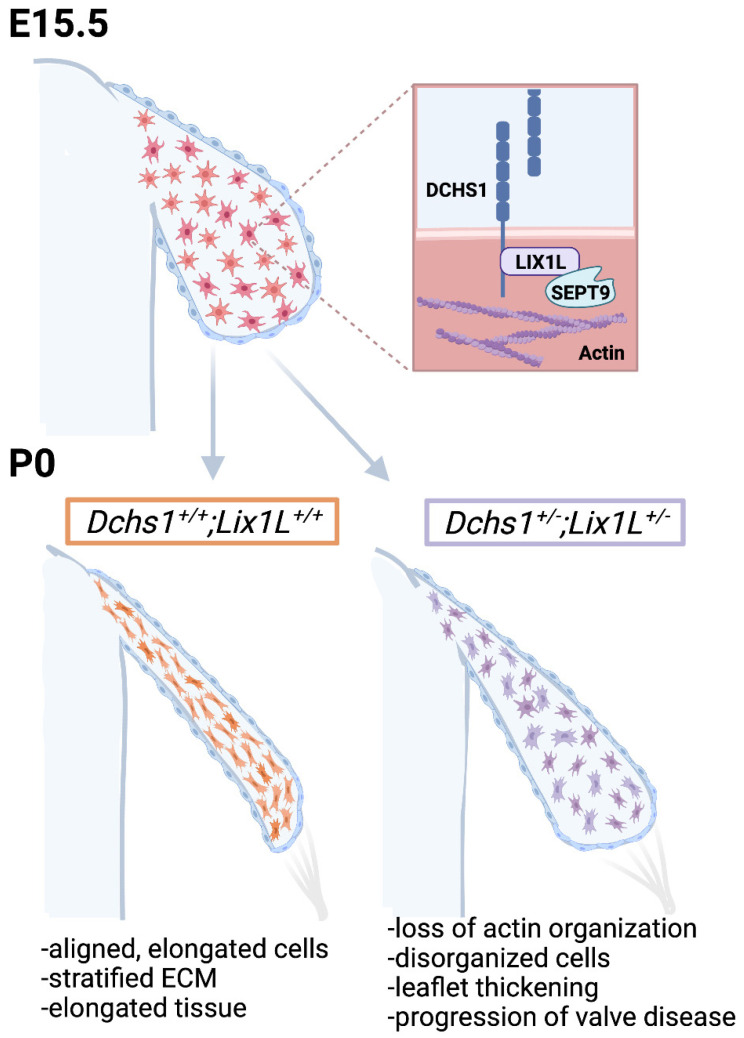
Mitral valve prolapse (MVP) is a common cardiac valve disease that often progresses to serious secondary complications requiring surgery. MVP manifests as extracellular matrix disorganization and biomechanically incompetent tissues in the adult setting. However, MVP has recently been shown to have a developmental basis, as multiple causal genes expressed during embryonic development have been identified. Disease phenotypes have been observed in mouse models with human MVP mutations as early as birth. This study focuses on the developmental function of DCHS1, one of the first genes to be shown as causal in multiple families with non-syndromic MVP. By using various biochemical techniques as well as mouse and cell culture models, we demonstrate a unique link between DCHS1-based cell adhesions and the septin-actin cytoskeleton through interactions with cytoplasmic protein Lix1-Like (LIX1L). This DCHS1-LIX1L-SEPT9 axis interacts with and promotes filamentous actin organization to direct cell-ECM alignment and valve tissue shape.
Keywords: DCHS1; cytoskeleton; heart valve development; mitral valve prolapse; septin.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Mutations in DCHS1 cause mitral valve prolapse.Nature. 2015 Sep 3;525(7567):109-13. doi: 10.1038/nature14670. Epub 2015 Aug 10. Nature. 2015. PMID: 26258302 Free PMC article.
-
Report of a rare case of congenital mitral valve prolapse with chronic kidney disease--reconsidered genotype-phenotypic correlations.Mol Genet Genomic Med. 2021 Jan;9(1):e1558. doi: 10.1002/mgg3.1558. Epub 2020 Nov 22. Mol Genet Genomic Med. 2021. PMID: 33225636 Free PMC article.
-
Genetics of syndromic and non-syndromic mitral valve prolapse.Heart. 2018 Jun;104(12):978-984. doi: 10.1136/heartjnl-2017-312420. Epub 2018 Jan 19. Heart. 2018. PMID: 29352010 Free PMC article. Review.
-
Genome-Wide Association Study-Driven Gene-Set Analyses, Genetic, and Functional Follow-Up Suggest GLIS1 as a Susceptibility Gene for Mitral Valve Prolapse.Circ Genom Precis Med. 2019 May;12(5):e002497. doi: 10.1161/CIRCGEN.119.002497. Circ Genom Precis Med. 2019. PMID: 31112420 Free PMC article.
-
Genetic background of mitral valve prolapse.Rev Cardiovasc Med. 2022 Mar 12;23(3):96. doi: 10.31083/j.rcm2303096. Rev Cardiovasc Med. 2022. PMID: 35345263 Review.
Cited by
-
Patient-derived induced pluripotent stem cells with a MERTK mutation exhibit cell junction abnormalities and aberrant cellular differentiation potential.World J Stem Cells. 2024 May 26;16(5):512-524. doi: 10.4252/wjsc.v16.i5.512. World J Stem Cells. 2024. PMID: 38817331 Free PMC article.
-
Septins as membrane influencers: direct play or in association with other cytoskeleton partners.Front Cell Dev Biol. 2023 Feb 17;11:1112319. doi: 10.3389/fcell.2023.1112319. eCollection 2023. Front Cell Dev Biol. 2023. PMID: 36875762 Free PMC article. Review.
-
Differential Development of the Chordae Tendineae and Anterior Leaflet of the Bovine Mitral Valve.J Cardiovasc Dev Dis. 2024 Mar 29;11(4):106. doi: 10.3390/jcdd11040106. J Cardiovasc Dev Dis. 2024. PMID: 38667724 Free PMC article.
-
Neuronal hyperactivity in neurons derived from individuals with gray matter heterotopia.Nat Commun. 2025 Feb 18;16(1):1737. doi: 10.1038/s41467-025-56998-1. Nat Commun. 2025. PMID: 39966398 Free PMC article.
-
Non-syndromal mitral valve prolapse (MVP): a common entity, but not commonly associated with DCHS1 or FLNA mutations.J Thorac Dis. 2022 Jun;14(6):2440-2442. doi: 10.21037/jtd-22-173. J Thorac Dis. 2022. PMID: 35813742 Free PMC article. No abstract available.
References
-
- Gammie J.S., Chikwe J., Badhwar V., Thibault D.P., Vemulapalli S., Thourani V.H., Gillinov M., Adams D.H., Rankin J.S., Ghoreishi M., et al. Isolated Mitral Valve Surgery: The Society of Thoracic Surgeons Adult Cardiac Surgery Database Analysis. Ann. Thorac. Surg. 2018;106:716–727. doi: 10.1016/j.athoracsur.2018.03.086. - DOI - PubMed
-
- Grigioni F., Tribouilloy C., Avierinos J.F., Barbieri A., Ferlito M., Trojette F., Tafanelli L., Branzi A., Szymanski C., Habib G., et al. Outcomes in Mitral Regurgitation Due to Flail Leaflets: A Multicenter European Study. JACC Cardiovasc. Imaging. 2008;1:133–141. doi: 10.1016/j.jcmg.2007.12.005. - DOI - PubMed
-
- Clemenceau A., Bérubé J.-C., Bélanger P., Gaudreault N., Lamontagne M., Toubal O., Clavel M.-A., Capoulade R., Mathieu P., Pibarot P., et al. Deleterious variants in DCHS1 are prevalent in sporadic cases of mitral valve prolapse. Mol. Genet. Genom. Med. 2017;6:114–120. doi: 10.1002/mgg3.347. - DOI - PMC - PubMed
Grants and funding
- R01 HL149696/HL/NHLBI NIH HHS/United States
- F31 HL152494/HL/NHLBI NIH HHS/United States
- C06 RR018823/RR/NCRR NIH HHS/United States
- HL131546 (R.A.N), GM103444 (R.A.N), HL149696 (R.A.N); HL122906 (R.A.N), T32HL007260 (T.B., C.G.), F31HL158243 (T.B.) F31152494 (KM), F31HL142159(DF), T32GM132055 (C. G.)/NH/NIH HHS/United States
- P20 GM103444/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
