

Angiotensin-converting enzyme gates brain circuit-specific plasticity via an endogenous opioid
- PMID: 35201898
- PMCID: PMC9233526
- DOI: 10.1126/science.abl5130
Angiotensin-converting enzyme gates brain circuit-specific plasticity via an endogenous opioid
Abstract
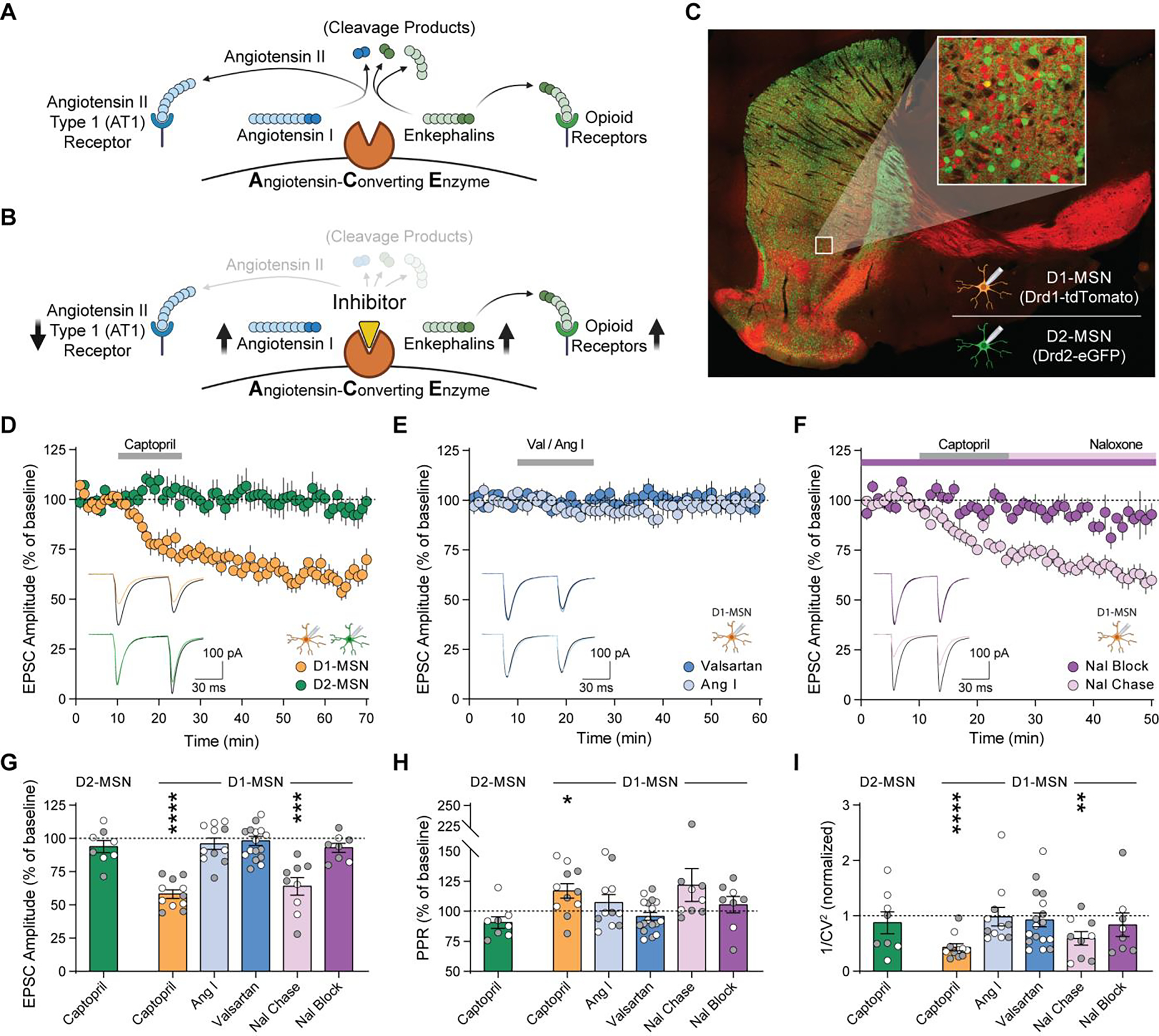
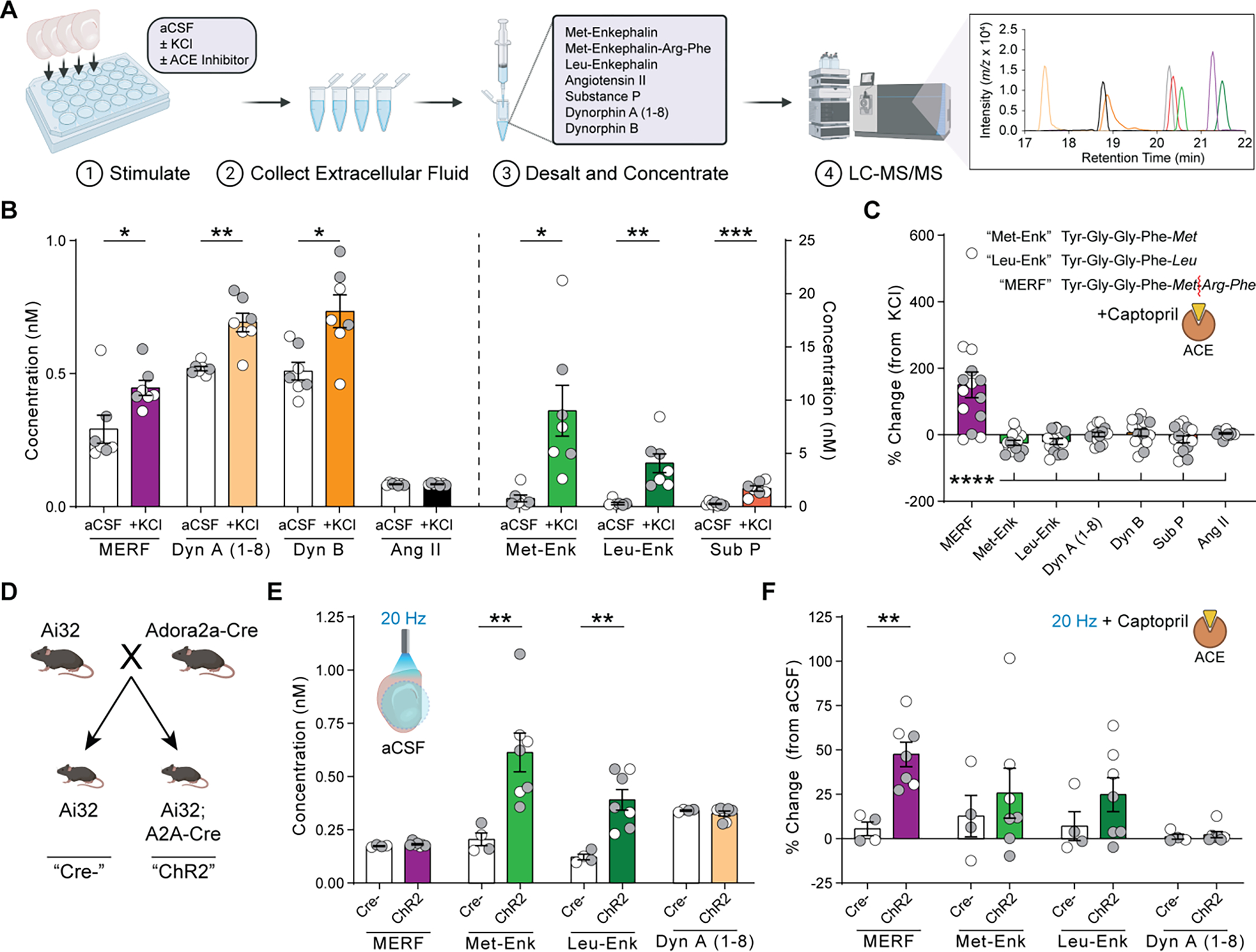
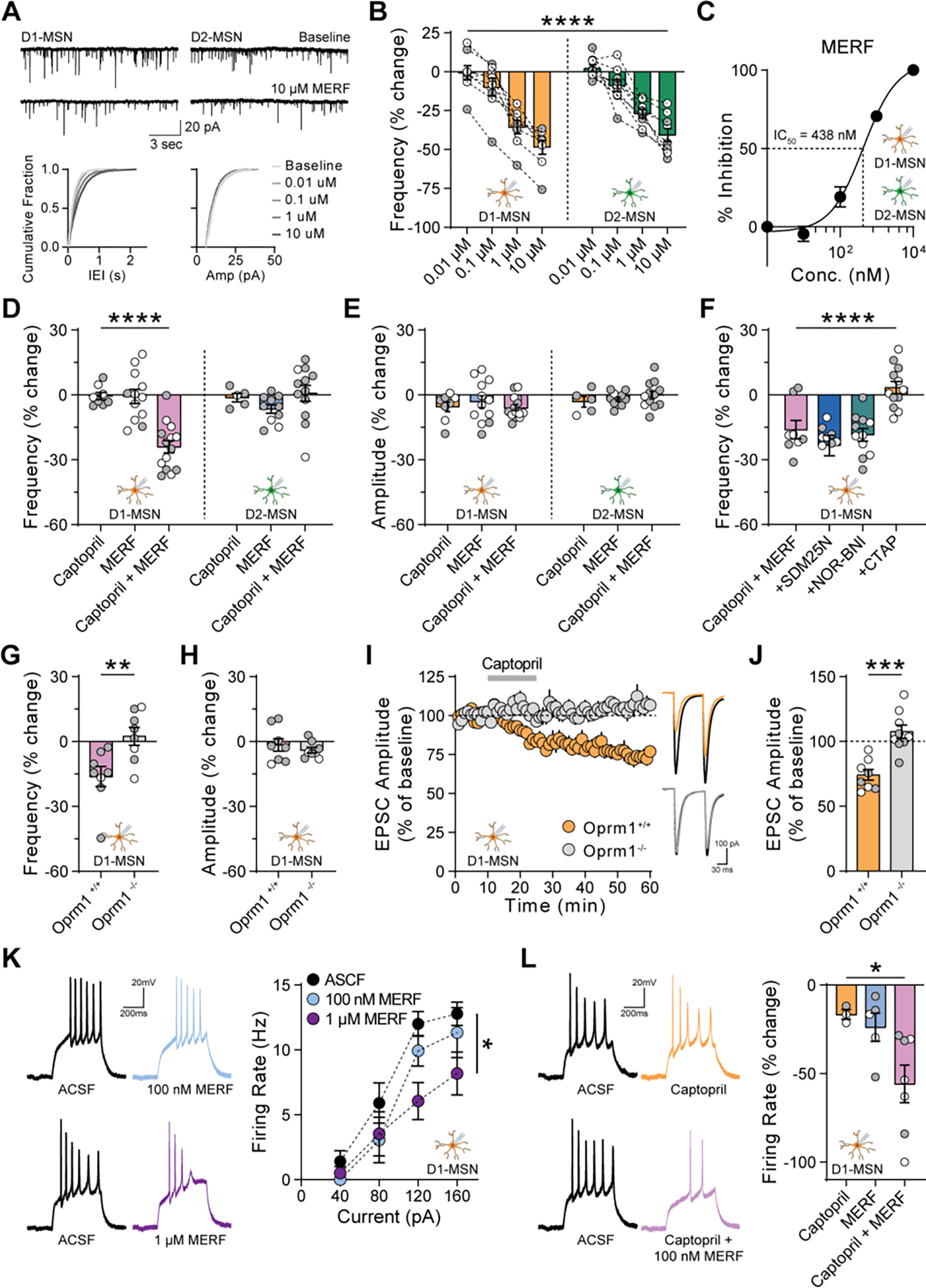
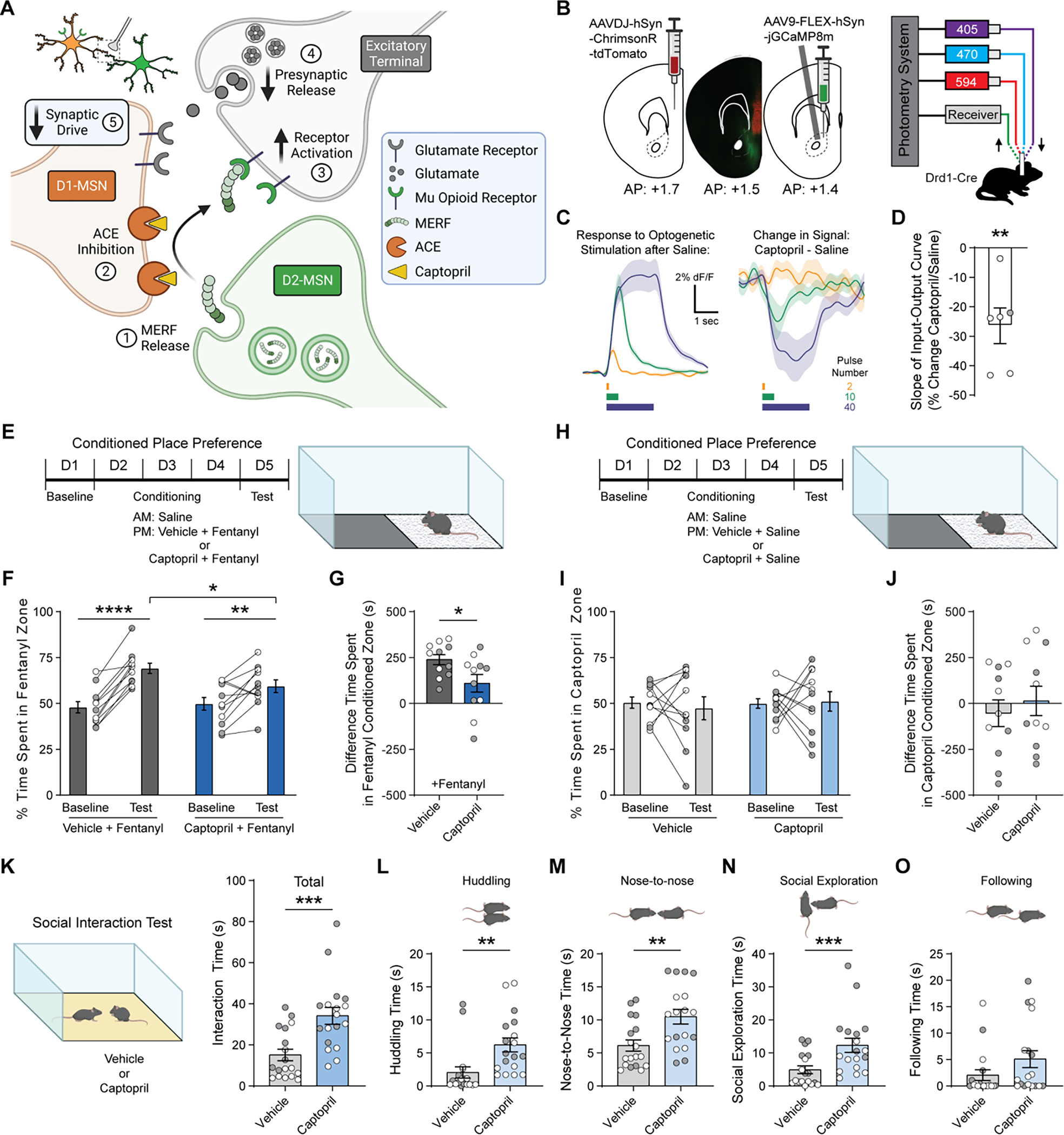
Angiotensin-converting enzyme (ACE) regulates blood pressure by cleaving angiotensin I to produce angiotensin II. In the brain, ACE is especially abundant in striatal tissue, but the function of ACE in striatal circuits remains poorly understood. We found that ACE degrades an unconventional enkephalin heptapeptide, Met-enkephalin-Arg-Phe, in the nucleus accumbens of mice. ACE inhibition enhanced µ-opioid receptor activation by Met-enkephalin-Arg-Phe, causing a cell type-specific long-term depression of glutamate release onto medium spiny projection neurons expressing the Drd1 dopamine receptor. Systemic ACE inhibition was not intrinsically rewarding, but it led to a decrease in conditioned place preference caused by fentanyl administration and an enhancement of reciprocal social interaction. Our results raise the enticing prospect that central ACE inhibition can boost endogenous opioid signaling for clinical benefit while mitigating the risk of addiction.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
