

CADASIL mutations sensitize the brain to ischemia via spreading depolarizations and abnormal extracellular potassium homeostasis
- PMID: 35202003
- PMCID: PMC9012276
- DOI: 10.1172/JCI149759
CADASIL mutations sensitize the brain to ischemia via spreading depolarizations and abnormal extracellular potassium homeostasis
Abstract
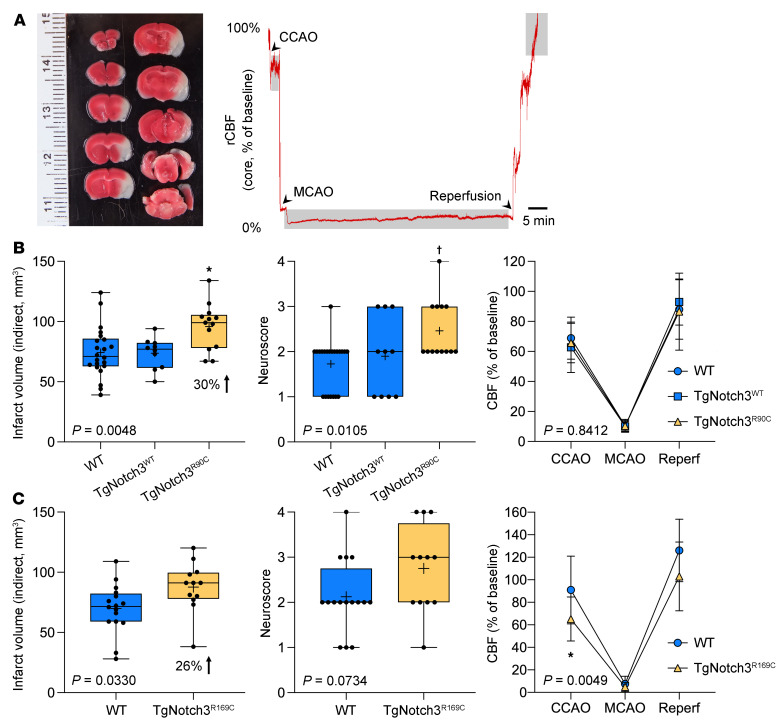
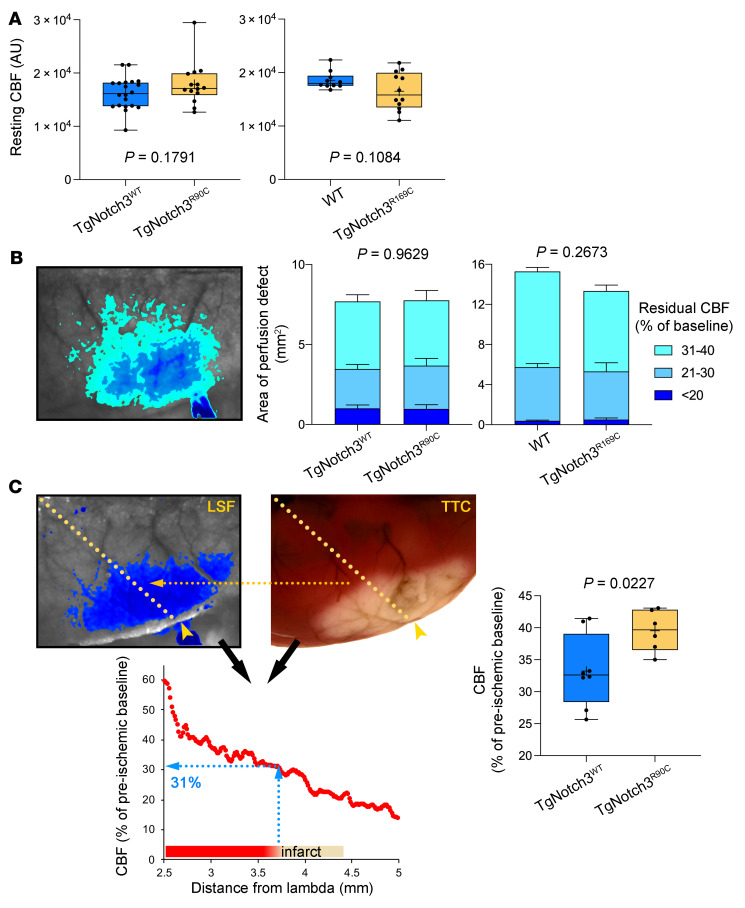
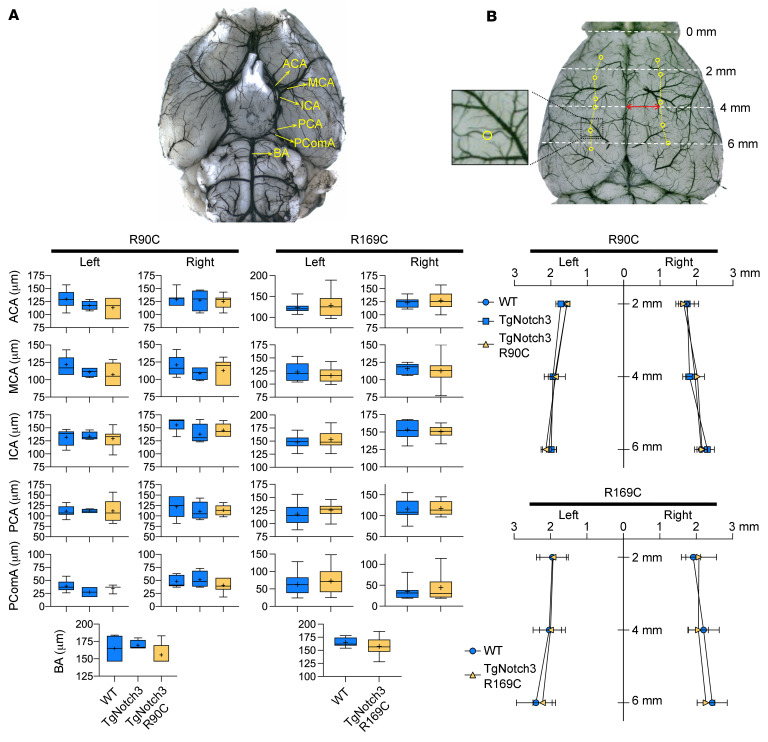
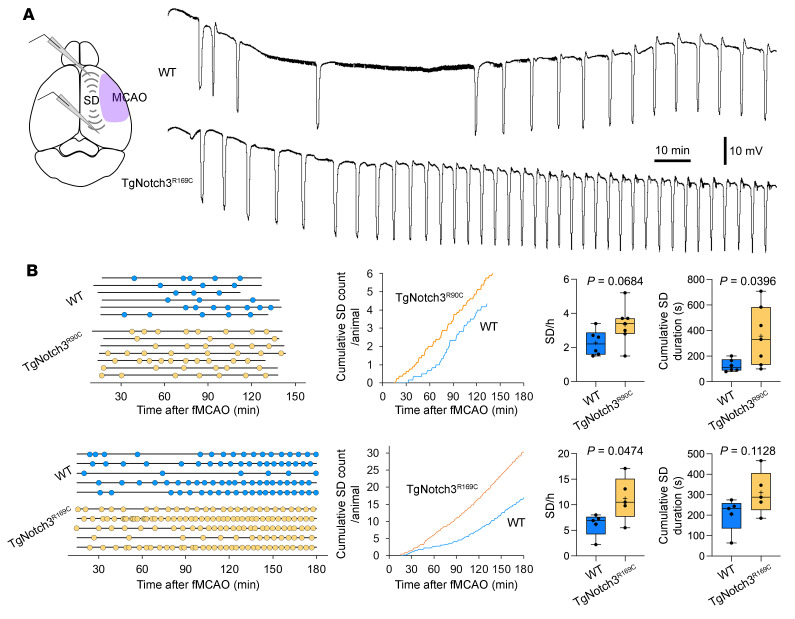
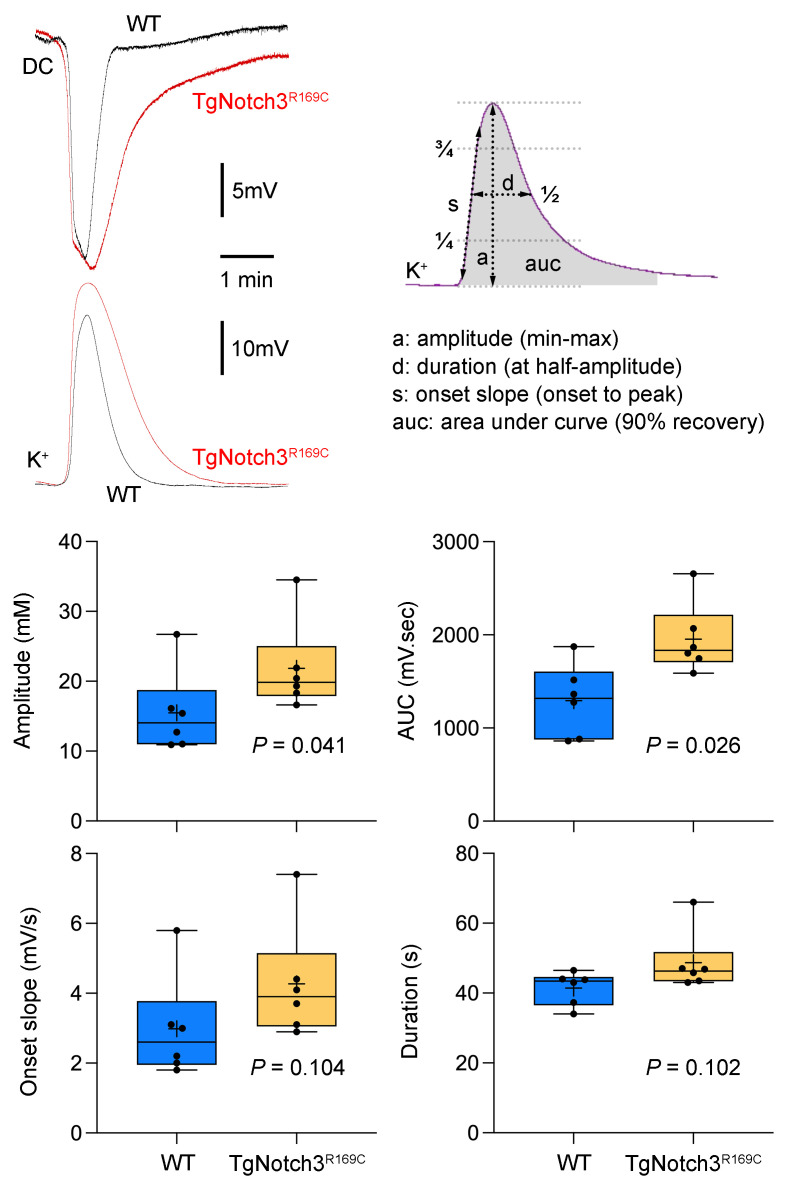
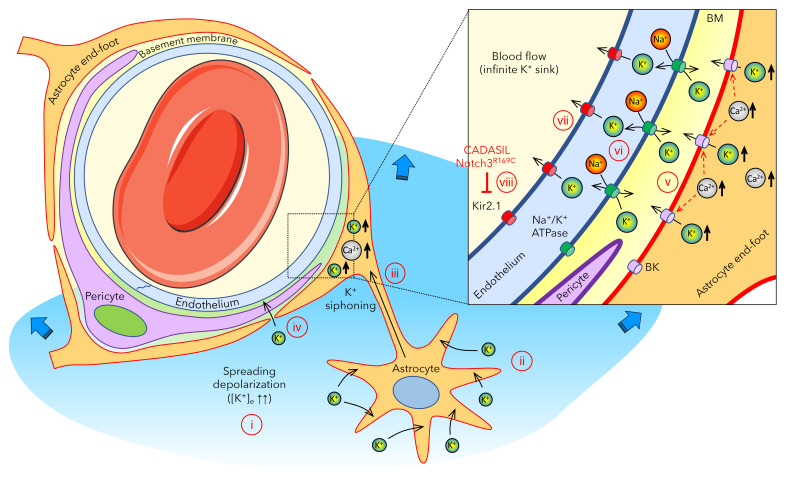
Cerebral autosomal dominant arteriopathy, subcortical infarcts, and leukoencephalopathy (CADASIL) is the most common monogenic form of small vessel disease characterized by migraine with aura, leukoaraiosis, strokes, and dementia. CADASIL mutations cause cerebrovascular dysfunction in both animal models and humans. Here, we showed that 2 different human CADASIL mutations (Notch3 R90C or R169C) worsen ischemic stroke outcomes in transgenic mice; this was explained by the higher blood flow threshold to maintain tissue viability compared with that in wild type (WT) mice. Both mutants developed larger infarcts and worse neurological deficits compared with WT mice, regardless of age or sex after filament middle cerebral artery occlusion. However, full-field laser speckle flowmetry during distal middle cerebral artery occlusion showed comparable perfusion deficits in mutants and their respective WT controls. Circle of Willis anatomy and pial collateralization also did not differ among the genotypes. In contrast, mutants had a higher cerebral blood flow threshold, below which infarction ensued, suggesting increased sensitivity of brain tissue to ischemia. Electrophysiological recordings revealed a 1.5- to 2-fold higher frequency of peri-infarct spreading depolarizations in CADASIL mutants. Higher extracellular K+ elevations during spreading depolarizations in the mutants implicated a defect in extracellular K+ clearance. Altogether, these data reveal a mechanism of enhanced vulnerability to ischemic injury linked to abnormal extracellular ion homeostasis and susceptibility to ischemic depolarizations in CADASIL.
Keywords: Microcirculation; Neuroscience; Potassium channels; Stroke.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
