

A new form of axonal pathology in a spinal model of neuromyelitis optica
- PMID: 35202467
- PMCID: PMC9166560
- DOI: 10.1093/brain/awac079
A new form of axonal pathology in a spinal model of neuromyelitis optica
Abstract
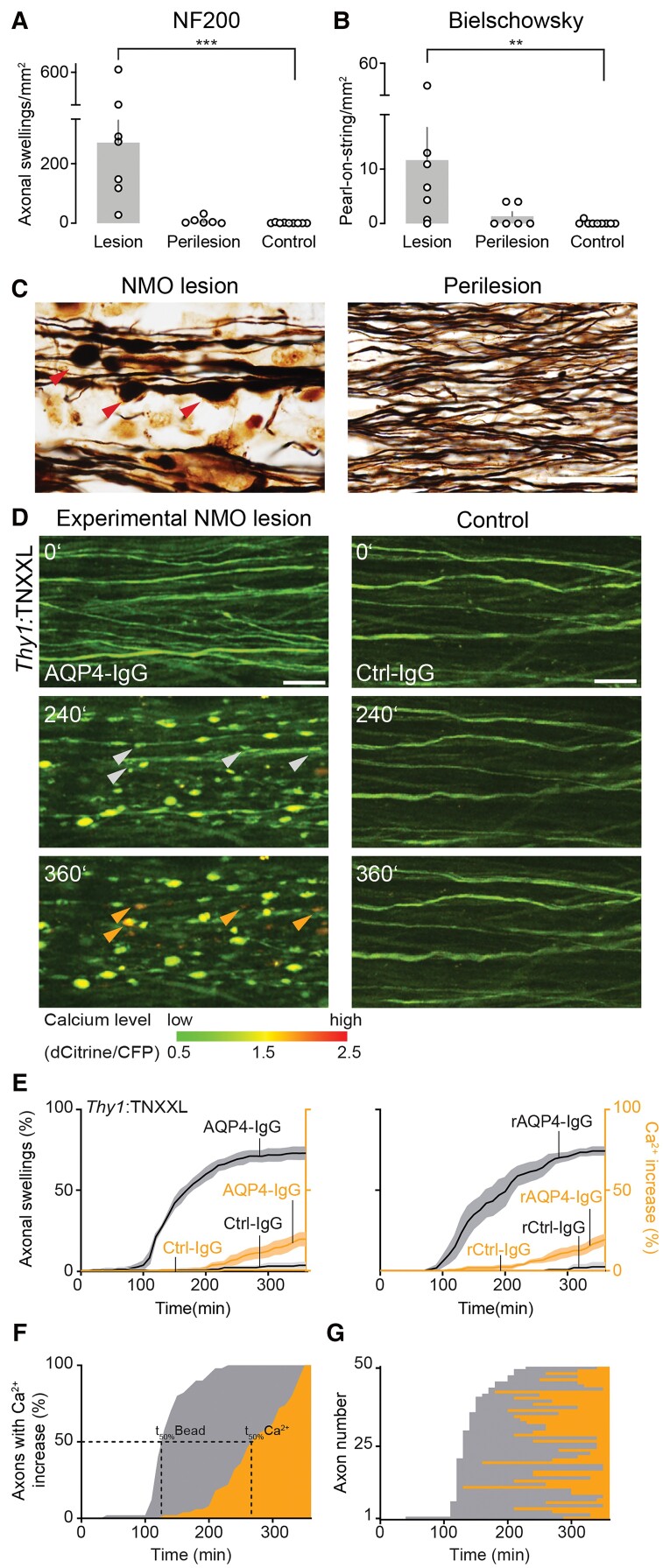
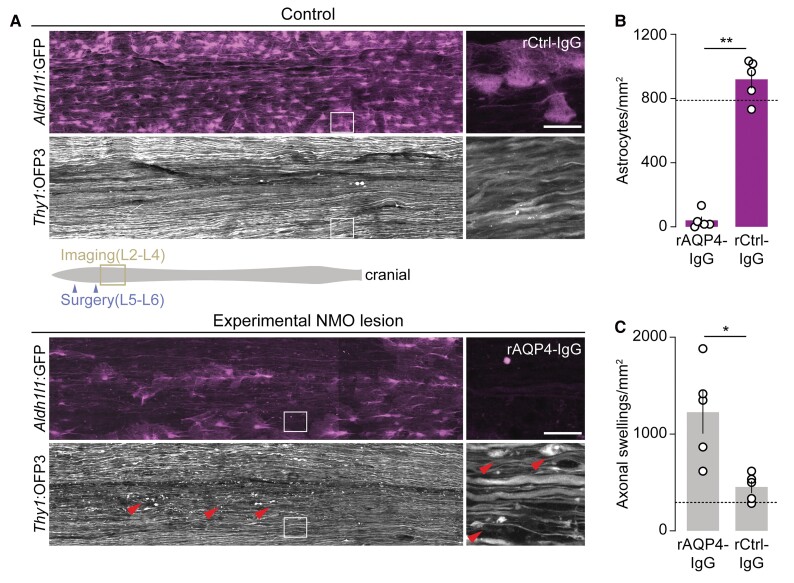
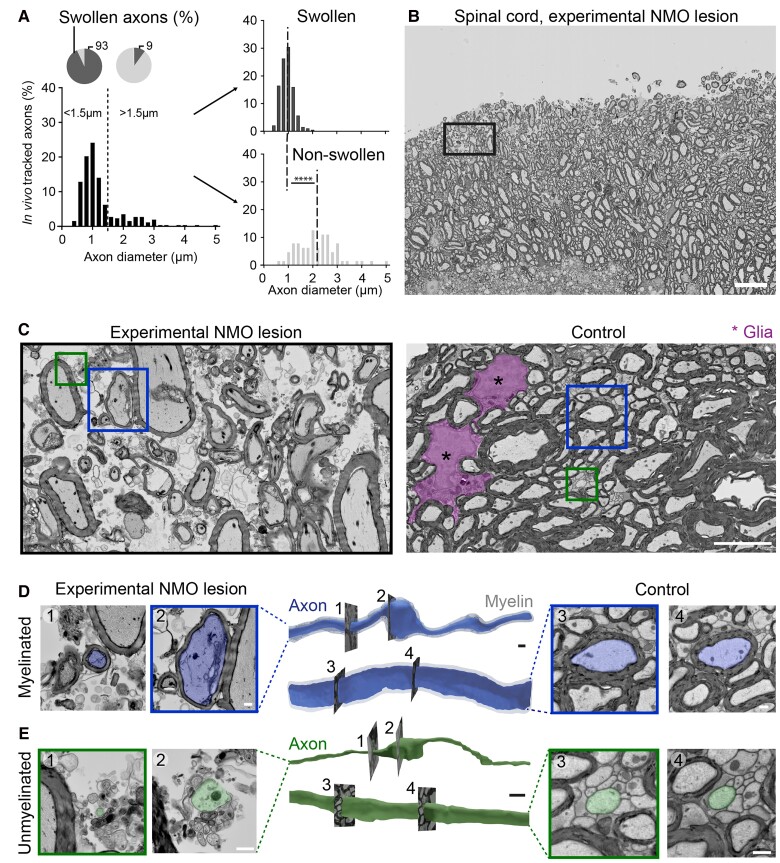
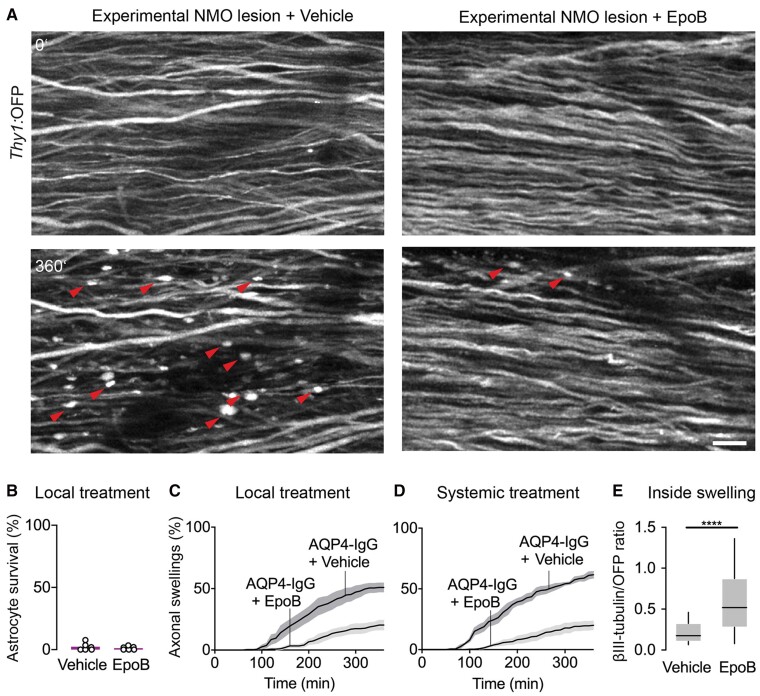
Neuromyelitis optica is a chronic neuroinflammatory disease, which primarily targets astrocytes and often results in severe axon injury of unknown mechanism. Neuromyelitis optica patients harbour autoantibodies against the astrocytic water channel protein, aquaporin-4 (AQP4-IgG), which induce complement-mediated astrocyte lysis and subsequent axon damage. Using spinal in vivo imaging in a mouse model of such astrocytopathic lesions, we explored the mechanism underlying neuromyelitis optica-related axon injury. Many axons showed a swift and morphologically distinct 'pearls-on-string' transformation also readily detectable in human neuromyelitis optica lesions, which especially affected small calibre axons independently of myelination. Functional imaging revealed that calcium homeostasis was initially preserved in this 'acute axonal beading' state, ruling out disruption of the axonal membrane, which sets this form of axon injury apart from previously described forms of traumatic and inflammatory axon damage. Morphological, pharmacological and genetic analyses showed that AQP4-IgG-induced axon injury involved osmotic stress and ionic overload, but does not appear to use canonical pathways of Wallerian-like degeneration. Subcellular analysis demonstrated remodelling of the axonal cytoskeleton in beaded axons, especially local loss of microtubules. Treatment with the microtubule stabilizer epothilone, a putative therapy approach for traumatic and degenerative axonopathies, prevented axonal beading, while destabilizing microtubules sensitized axons for beading. Our results reveal a distinct form of immune-mediated axon pathology in neuromyelitis optica that mechanistically differs from known cascades of post-traumatic and inflammatory axon loss, and suggest a new strategy for neuroprotection in neuromyelitis optica and related diseases.
Keywords: aquaporin-4; astrocytopathy; neurodegeneration; neuroinflammation; neuromyelitis optica.
© The Author(s) 2022. Published by Oxford University Press on behalf of the Guarantors of Brain.
Figures

Comment in
-
From astrocyte destruction to axon injury: watching lesion evolution in experimental neuromyelitis optica.Brain. 2022 Jun 3;145(5):1581-1583. doi: 10.1093/brain/awac141. Brain. 2022. PMID: 35438768 No abstract available.
References
-
- Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosis. Lancet. 2004;364:2106–2112. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
