

Channelopathy Genes in Pulmonary Arterial Hypertension
- PMID: 35204766
- PMCID: PMC8961593
- DOI: 10.3390/biom12020265
Channelopathy Genes in Pulmonary Arterial Hypertension
Abstract
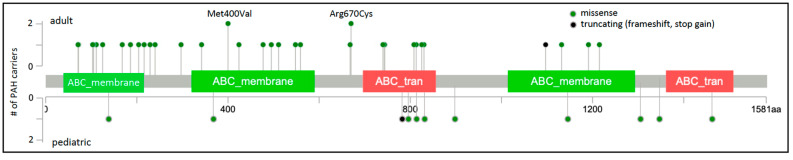
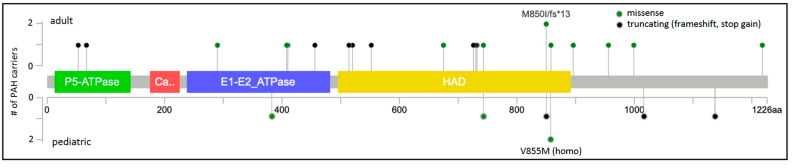
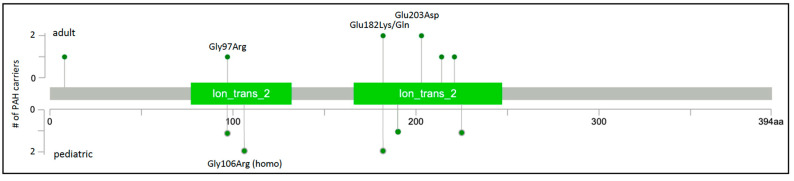
Pulmonary arterial hypertension (PAH) is a rare, progressive vasculopathy with significant cardiopulmonary morbidity and mortality. The underlying pathogenetic mechanisms are heterogeneous and current therapies aim to decrease pulmonary vascular resistance but no curative treatments are available. Causal genetic variants can be identified in ~13% of adults and 43% of children with PAH. Knowledge of genetic diagnoses can inform clinical management of PAH, including multimodal medical treatment, surgical intervention and transplantation decisions, and screening for associated conditions, as well as risk stratification for family members. Roles for rare variants in three channelopathy genes-ABCC8, ATP13A3, and KCNK3-have been validated in multiple PAH cohorts, and in aggregate explain ~2.7% of PAH cases. Complete or partial loss of function has been demonstrated for PAH-associated variants in ABCC8 and KCNK3. Channels can be excellent targets for drugs, and knowledge of mechanisms for channel mutations may provide an opportunity for the development of PAH biomarkers and novel therapeutics for patients with hereditary PAH but also potentially more broadly for all patients with PAH.
Keywords: channelopathy; genetics; lung disease; pulmonary arterial hypertension.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Vonk-Noordegraaf A., Haddad F., Chin K.M., Forfia P.R., Kawut S.M., Lumens J., Naeije R., Newman J., Oudiz R.J., Provencher S., et al. Right heart adaptation to pulmonary arterial hypertension: Physiology and pathobiology. J. Am. Coll. Cardiol. 2013;62((Suppl. 25)):D22–D33. doi: 10.1016/j.jacc.2013.10.027. - DOI - PubMed
-
- Humbert M., Guignabert C., Bonnet S., Dorfmuller P., Klinger J.R., Nicolls M.R., Olschewski A.J., Pullamsetti S.S., Schermuly R.T., Stenmark K.R., et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019;53:1801887. doi: 10.1183/13993003.01887-2018. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
