

DRPPM-EASY: A Web-Based Framework for Integrative Analysis of Multi-Omics Cancer Datasets
- PMID: 35205126
- PMCID: PMC8869715
- DOI: 10.3390/biology11020260
DRPPM-EASY: A Web-Based Framework for Integrative Analysis of Multi-Omics Cancer Datasets
Abstract
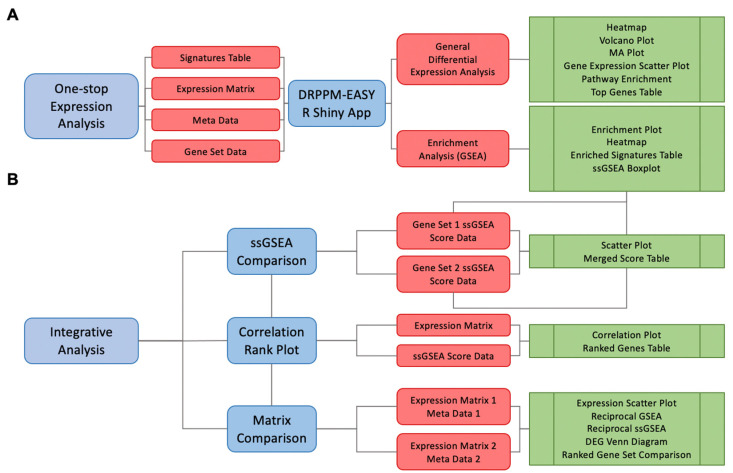
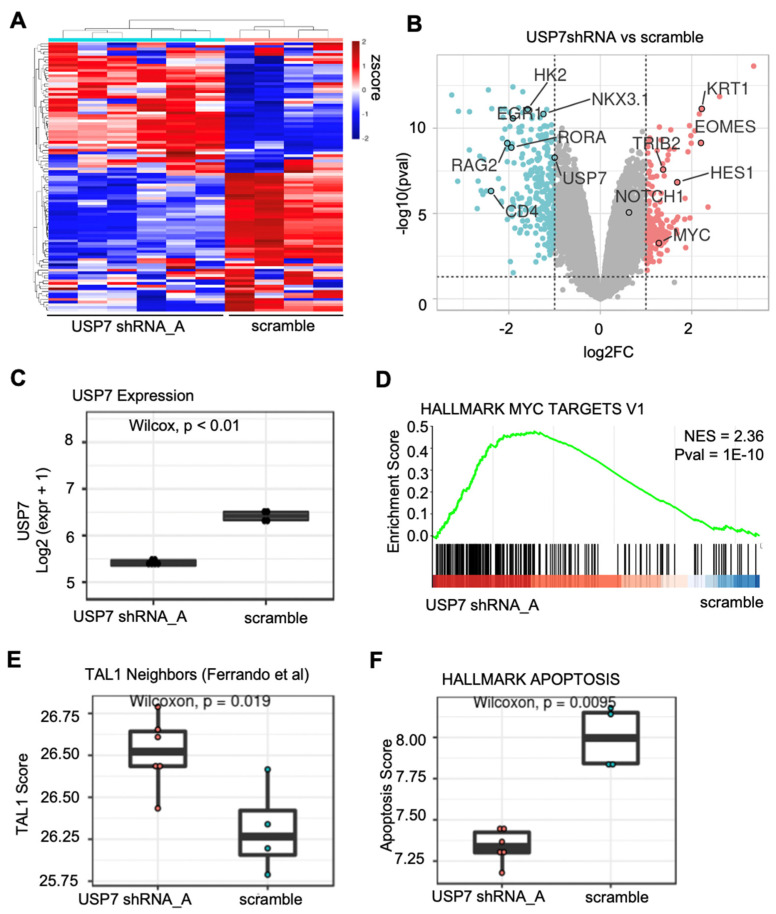
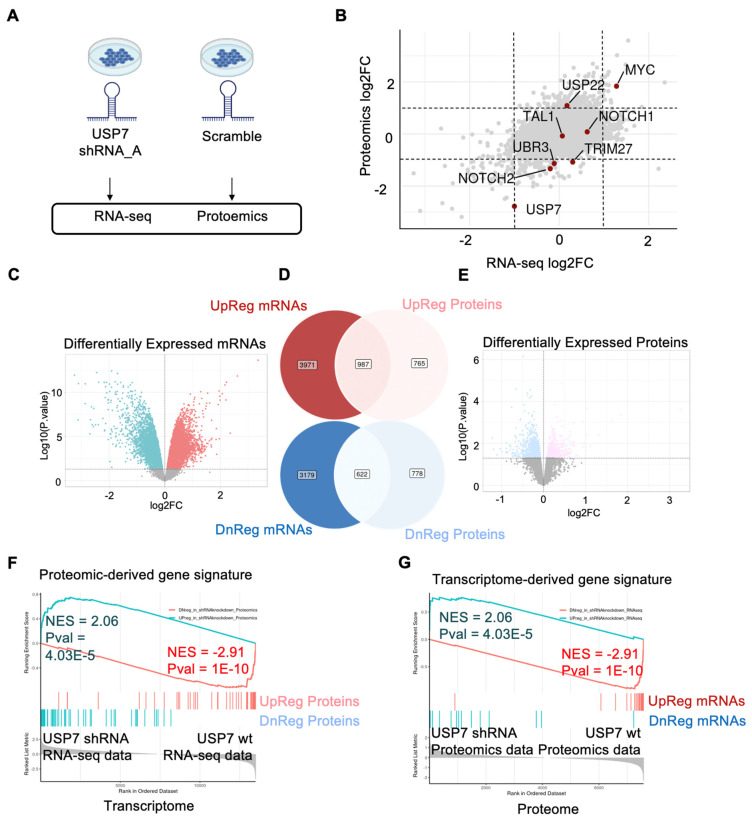
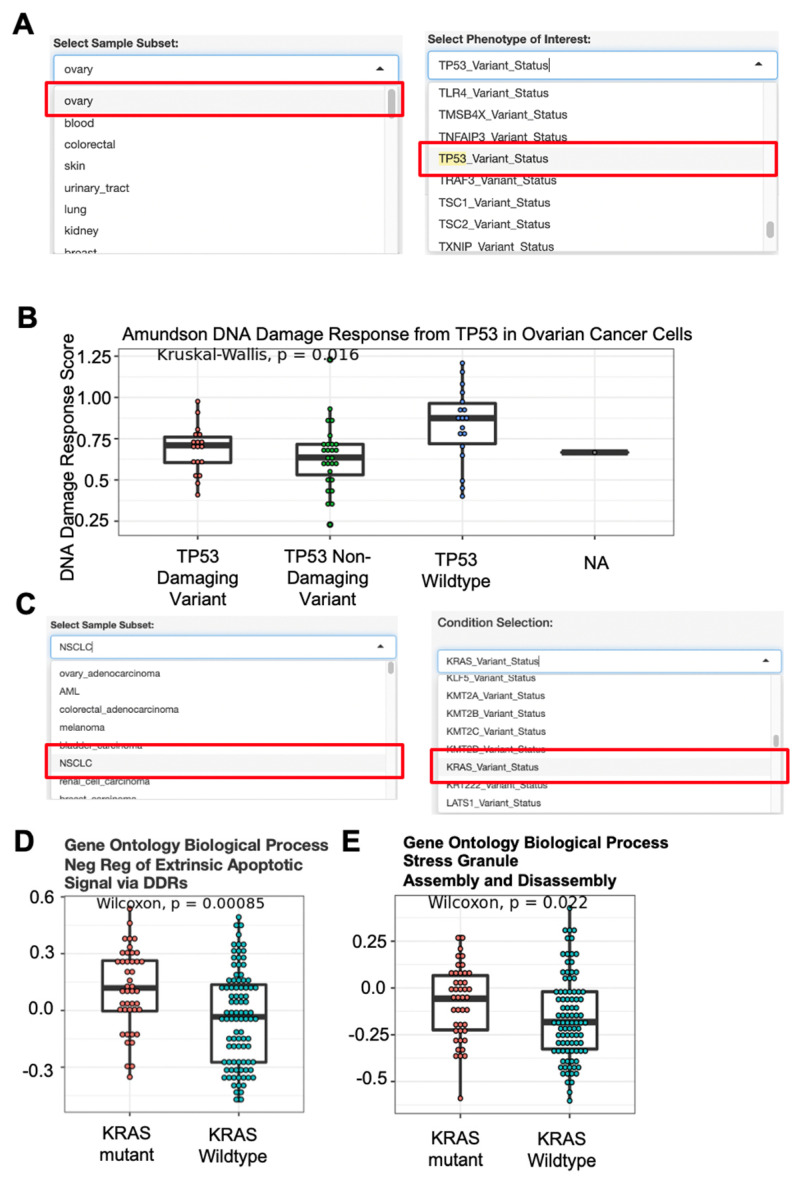
High-throughput transcriptomic and proteomic analyses are now routinely applied to study cancer biology. However, complex omics integration remains challenging and often time-consuming. Here, we developed DRPPM-EASY, an R Shiny framework for integrative multi-omics analysis. We applied our application to analyze RNA-seq data generated from a USP7 knockdown in T-cell acute lymphoblastic leukemia (T-ALL) cell line, which identified upregulated expression of a TAL1-associated proliferative signature in T-cell acute lymphoblastic leukemia cell lines. Next, we performed proteomic profiling of the USP7 knockdown samples. Through DRPPM-EASY-Integration, we performed a concurrent analysis of the transcriptome and proteome and identified consistent disruption of the protein degradation machinery and spliceosome in samples with USP7 silencing. To further illustrate the utility of the R Shiny framework, we developed DRPPM-EASY-CCLE, a Shiny extension preloaded with the Cancer Cell Line Encyclopedia (CCLE) data. The DRPPM-EASY-CCLE app facilitates the sample querying and phenotype assignment by incorporating meta information, such as genetic mutation, metastasis status, sex, and collection site. As proof of concept, we verified the expression of TP53 associated DNA damage signature in TP53 mutated ovary cancer cells. Altogether, our open-source application provides an easy-to-use framework for omics exploration and discovery.
Keywords: CCLE; R Shiny application; RNA-seq; T-cell acute lymphoblastic leukemia; multi-omics analysis; proteomics.
Conflict of interest statement
The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
