

The Difference in the Proportions of Deleterious Variations within and between Populations Influences the Estimation of FST
- PMID: 35205239
- PMCID: PMC8872184
- DOI: 10.3390/genes13020194
The Difference in the Proportions of Deleterious Variations within and between Populations Influences the Estimation of FST
Abstract
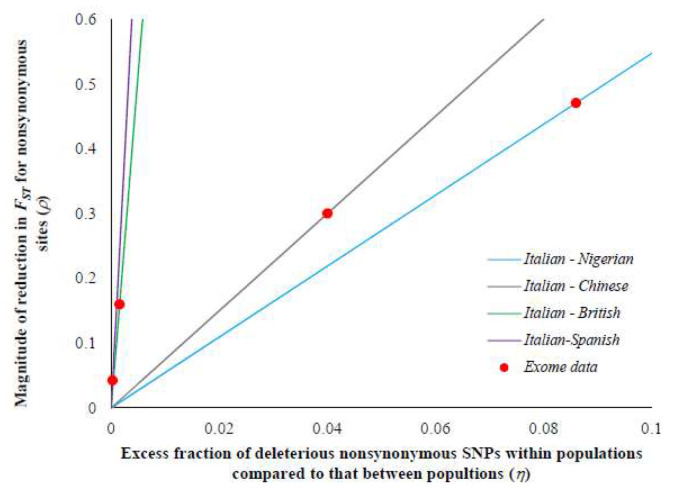
Estimating the extent of genetic differentiation between populations is an important measure in population genetics, ecology and evolutionary biology. The fixation index, or FST, is an important measure, which is routinely used to quantify this. Previous studies have shown that the FST estimated for selectively constrained regions was significantly lower than that estimated for neutral regions. By deriving the theoretical relationship between FST at neutral and constrained sites, we show that excess in the fraction of deleterious variations segregating within populations compared to those segregating between populations is the cause for the reduction in FST estimated at constrained sites. Using whole-genome data, our results revealed that the magnitude of reduction in FST estimates obtained for selectively constrained regions was much higher for distantly related populations compared to those estimated for closely related pairs. For example, the reduction was 47% for comparison between Europeans and Africans, 30% for the European and Asian comparison, 16% for the Northern and Southern European pair, and only 4% for the comparison involving two Southern European (Italian and Spanish) populations. Since deleterious variants are purged over time due to purifying selection, their contribution to the among-population diversity at constrained sites decreases with the increase in the divergence between populations. However, within-population diversities remain the same for all pairs compared; therefore, the FST estimated at constrained sites for distantly related populations are much smaller than those estimated for closely related populations. We obtained similar results when only the SNPs with similar allele frequencies at neutral and constrained sites were used. Our results suggest that the level of population divergence should be considered when comparing constrained site FST estimates from different pairs of populations.
Keywords: FST; deleterious mutations; population differentiation; temporal distributions and population genetics theory.
Conflict of interest statement
The authors declare no conflict of interest.
Figures




References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
