

Ischemic stroke causes Parkinson's disease-like pathology and symptoms in transgenic mice overexpressing alpha-synuclein
- PMID: 35209932
- PMCID: PMC8867857
- DOI: 10.1186/s40478-022-01327-6
Ischemic stroke causes Parkinson's disease-like pathology and symptoms in transgenic mice overexpressing alpha-synuclein
Abstract
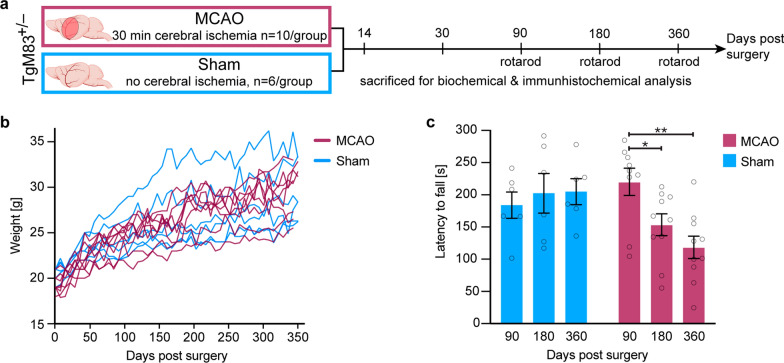
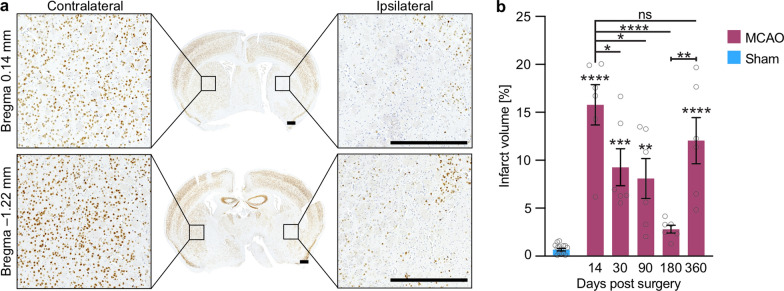
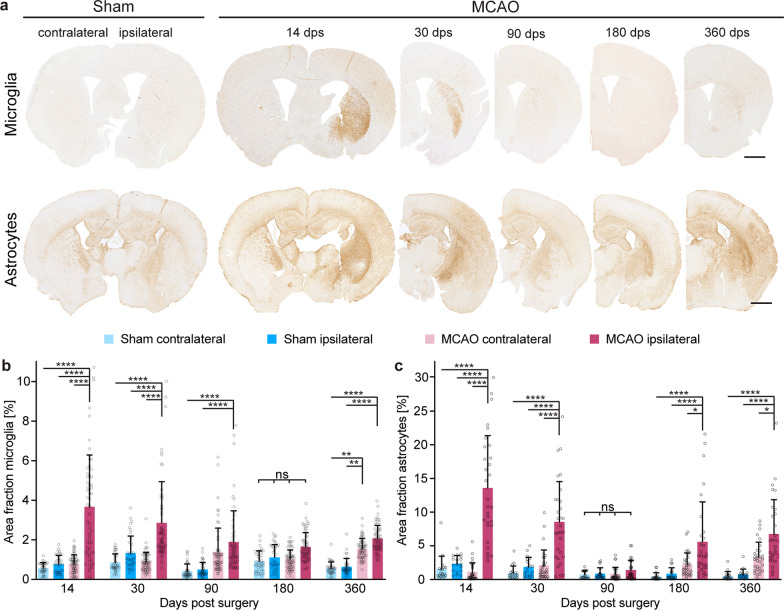
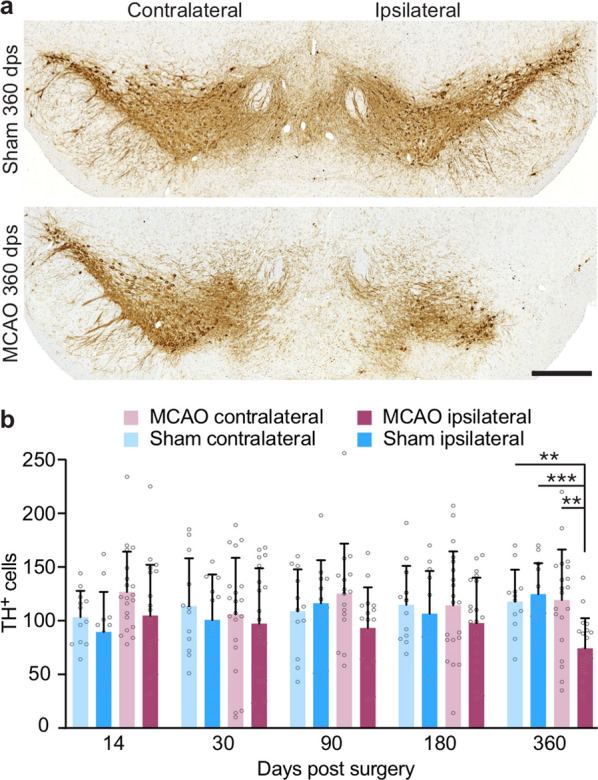
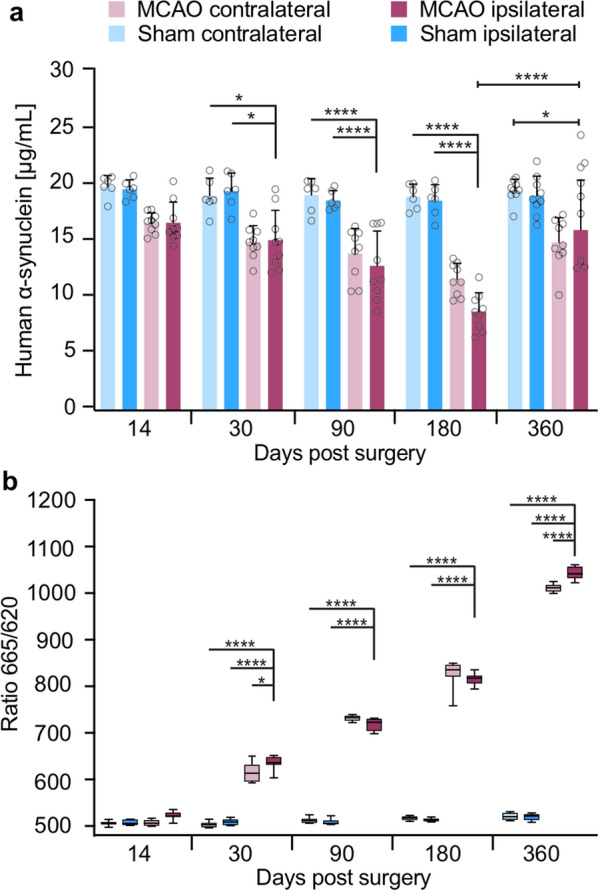
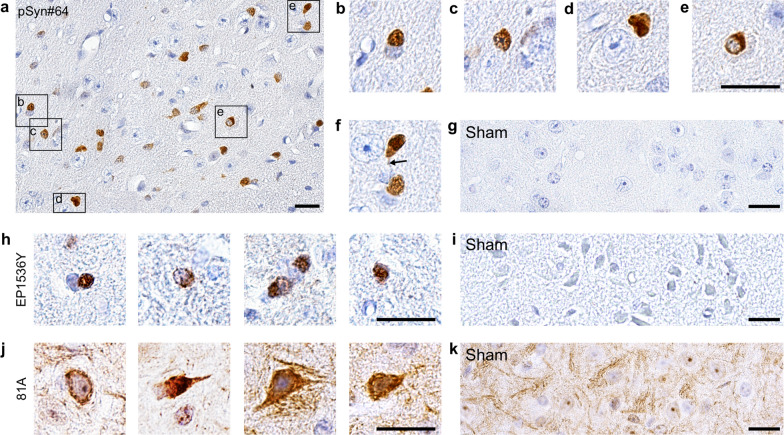
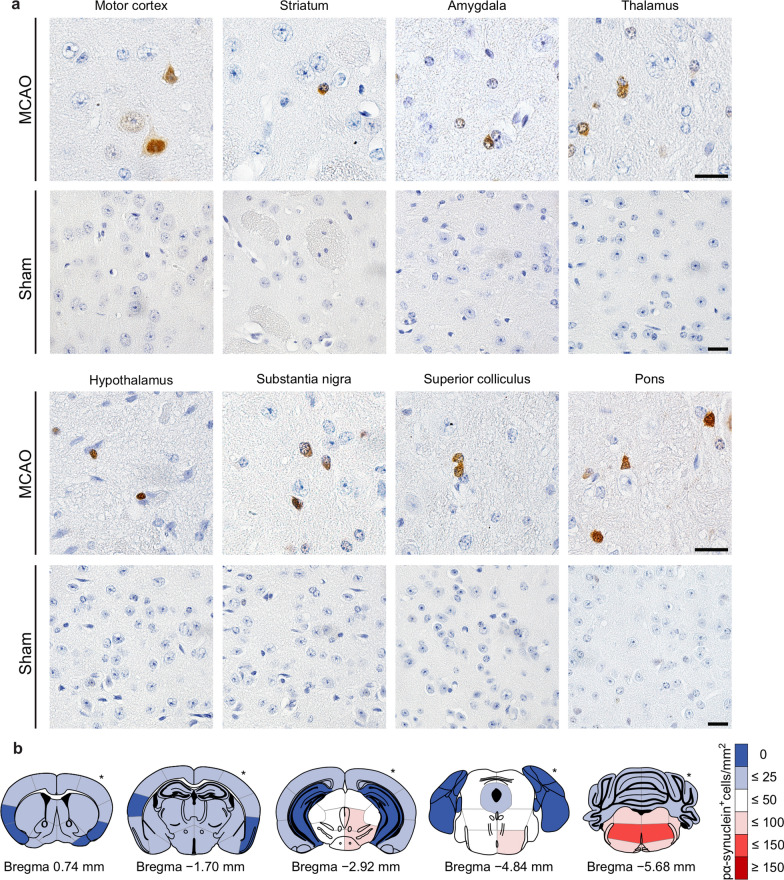
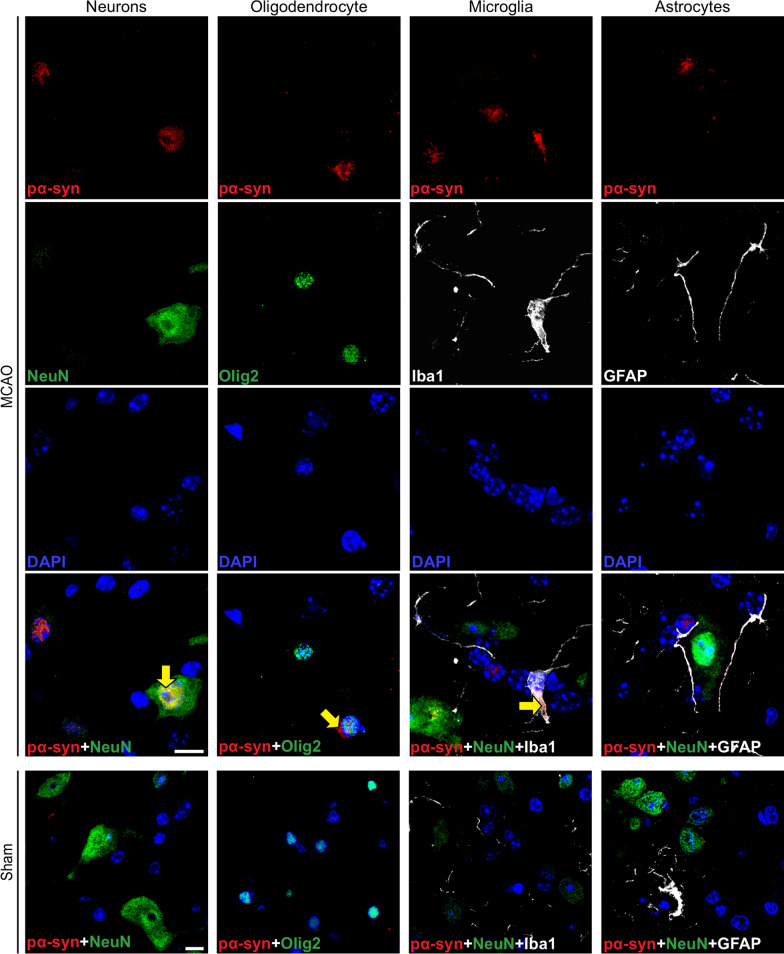
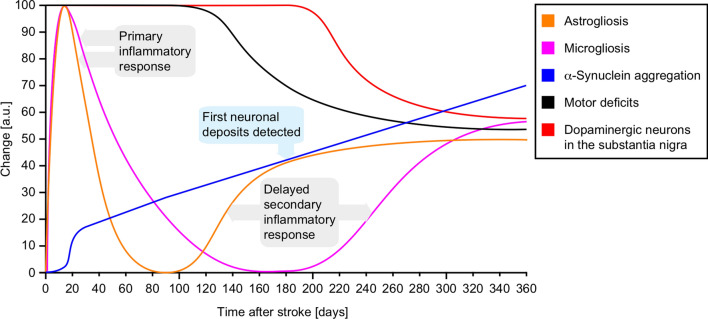
The etiology of Parkinson's disease is poorly understood and is most commonly associated with advancing age, genetic predisposition, or environmental toxins. Epidemiological findings suggest that patients have a higher risk of developing Parkinson's disease after ischemic stroke, but this potential causality lacks mechanistic evidence. We investigated the long-term effects of ischemic stroke on pathogenesis in hemizygous TgM83 mice, which express human α-synuclein with the familial A53T mutation without developing any neuropathology or signs of neurologic disease for more than 600 days. We induced transient focal ischemia by middle cerebral artery occlusion in 2-month-old TgM83+/- mice and monitored their behavior and health status for up to 360 days post surgery. Groups of mice were sacrificed at 14, 30, 90, 180, and 360 days after surgery for neuropathological analysis of their brains. Motor deficits first appeared 6 months after focal ischemia and worsened until 12 months afterward. Immunohistochemical analysis revealed ischemia-induced neuronal loss in the infarct region and astrogliosis and microgliosis indicative of an inflammatory response, which was most pronounced at 14 days post surgery. Infarct volume and inflammation gradually decreased in size and severity until 180 days post surgery. Surprisingly, neuronal loss and inflammation were increased again by 360 days post surgery. These changes were accompanied by a continuous increase in α-synuclein aggregation, its neuronal deposition, and a late loss of dopaminergic neurons in the substantia nigra, which we detected at 360 days post surgery. Control animals that underwent sham surgery without middle cerebral artery occlusion showed no signs of disease or neuropathology. Our results establish a mechanistic link between ischemic stroke and Parkinson's disease and provide an animal model for studying possible interventions.
Keywords: Alpha-synuclein; Ischemia; Ischemic stroke; Parkinson’s disease; Stroke; Synucleinopathy.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Dement A. Alzheimer's disease facts and figures. Alzheimer's Dementia. 2020;16:391–460. doi: 10.1002/alz.12068. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
