Comparative pangenome analysis of capsulated Haemophilus influenzae serotype f highlights their high genomic stability
- PMID: 35210526
- PMCID: PMC8873416
- DOI: 10.1038/s41598-022-07185-5
Comparative pangenome analysis of capsulated Haemophilus influenzae serotype f highlights their high genomic stability
Abstract
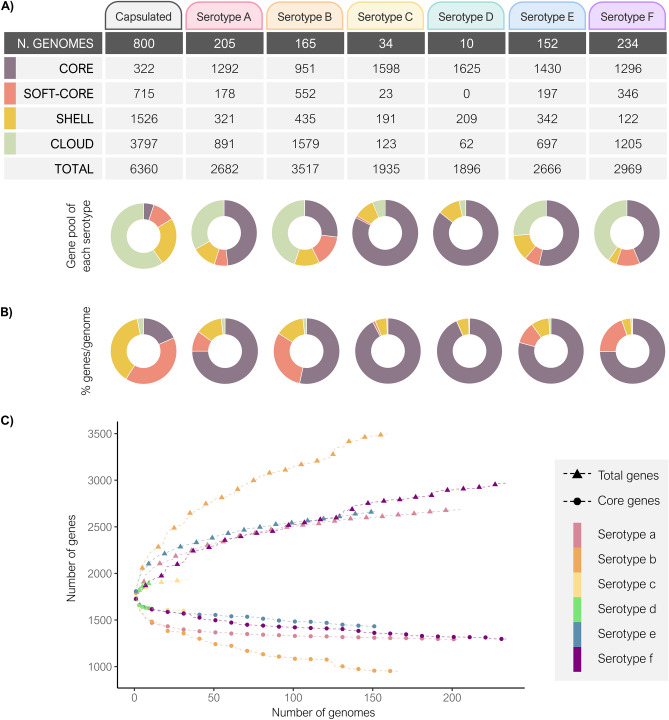
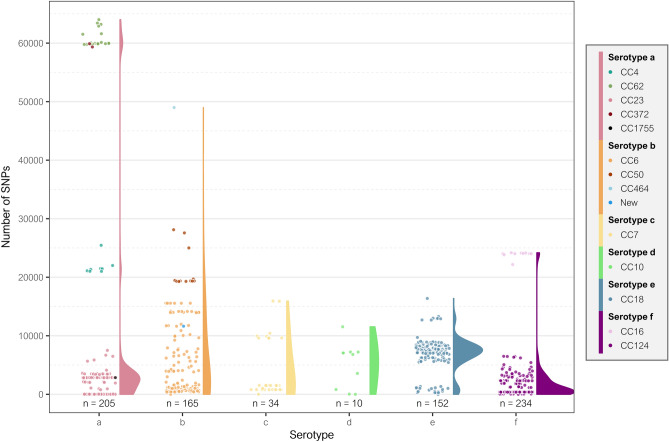
Haemophilus influenzae is an opportunistic pathogen adapted to the human respiratory tract. Non-typeable H. influenzae are highly heterogeneous, but few studies have analysed the genomic variability of capsulated strains. This study aims to examine the genetic diversity of 37 serotype f isolates from the Netherlands, Portugal, and Spain, and to compare all capsulated genomes available on public databases. Serotype f isolates belonged to CC124 and shared few single nucleotide polymorphisms (SNPs) (n = 10,999), but a high core genome (> 80%). Three main clades were identified by the presence of 75, 60 and 41 exclusive genes for each clade, respectively. Multi-locus sequence type analysis of all capsulated genomes revealed a reduced number of clonal complexes associated with each serotype. Pangenome analysis showed a large pool of genes (n = 6360), many of which were accessory genome (n = 5323). Phylogenetic analysis revealed that serotypes a, b, and f had greater diversity. The total number of SNPs in serotype f was significantly lower than in serotypes a, b, and e (p < 0.0001), indicating low variability within the serotype f clonal complexes. Capsulated H. influenzae are genetically homogeneous, with few lineages in each serotype. Serotype f has high genetic stability regardless of time and country of isolation.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures