

An amiloride derivative is active against the F1Fo-ATP synthase and cytochrome bd oxidase of Mycobacterium tuberculosis
- PMID: 35210534
- PMCID: PMC8873251
- DOI: 10.1038/s42003-022-03110-8
An amiloride derivative is active against the F1Fo-ATP synthase and cytochrome bd oxidase of Mycobacterium tuberculosis
Abstract
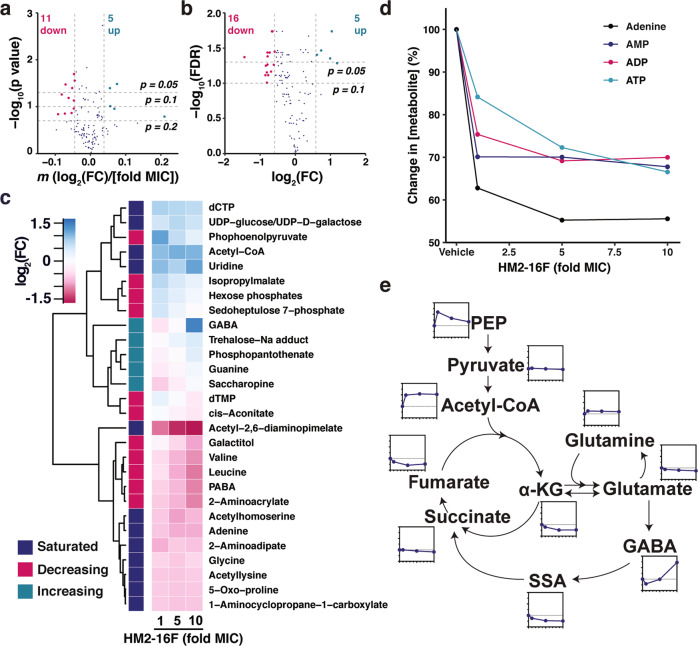
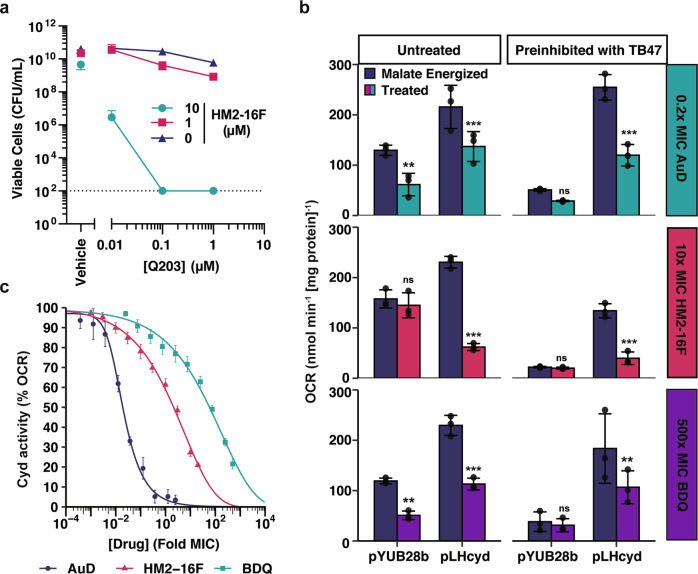
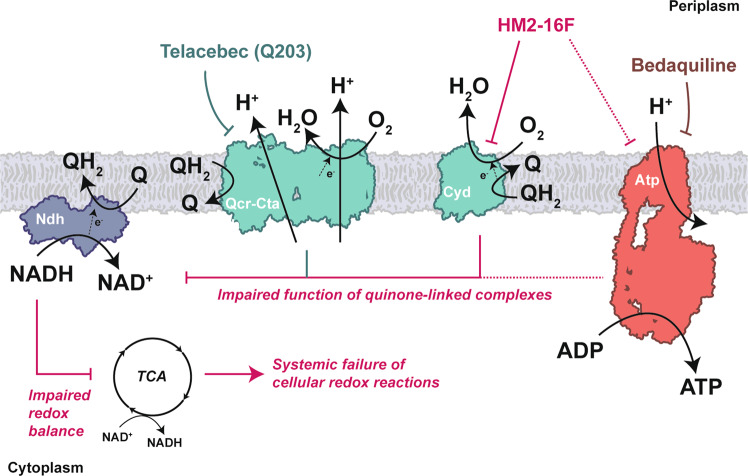
Increasing antimicrobial resistance compels the search for next-generation inhibitors with differing or multiple molecular targets. In this regard, energy conservation in Mycobacterium tuberculosis has been clinically validated as a promising new drug target for combatting drug-resistant strains of M. tuberculosis. Here, we show that HM2-16F, a 6-substituted derivative of the FDA-approved drug amiloride, is an anti-tubercular inhibitor with bactericidal properties comparable to the FDA-approved drug bedaquiline (BDQ; Sirturo®) and inhibits the growth of bedaquiline-resistant mutants. We show that HM2-16F weakly inhibits the F1Fo-ATP synthase, depletes ATP, and affects the entry of acetyl-CoA into the Krebs cycle. HM2-16F synergizes with the cytochrome bcc-aa3 oxidase inhibitor Q203 (Telacebec) and co-administration with Q203 sterilizes in vitro cultures in 14 days. Synergy with Q203 occurs via direct inhibition of the cytochrome bd oxidase by HM2-16F. This study shows that amiloride derivatives represent a promising discovery platform for targeting energy generation in drug-resistant tuberculosis.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
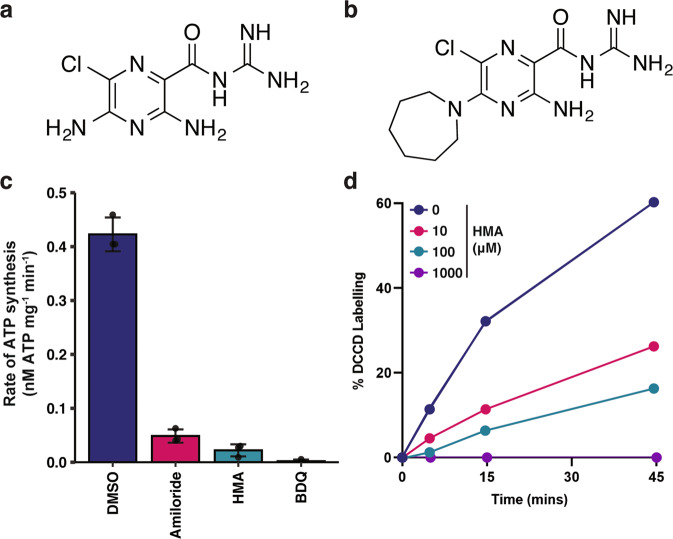
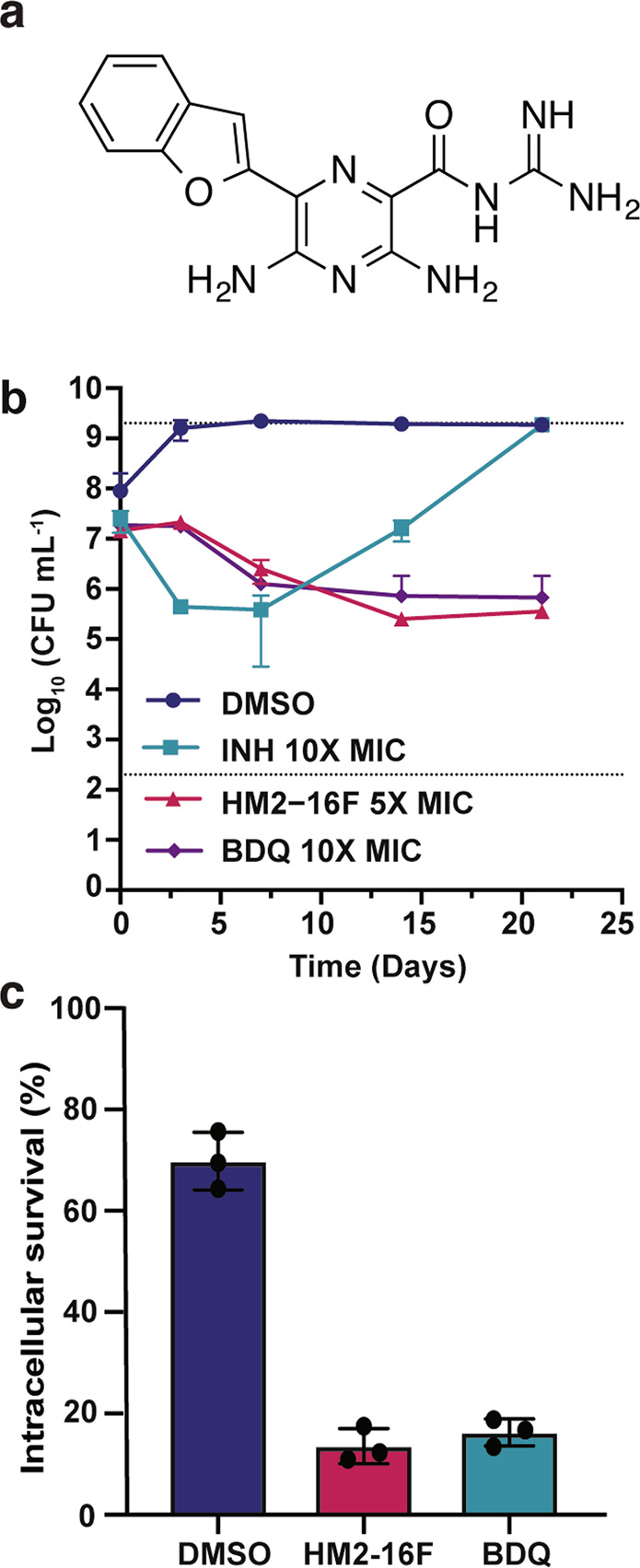
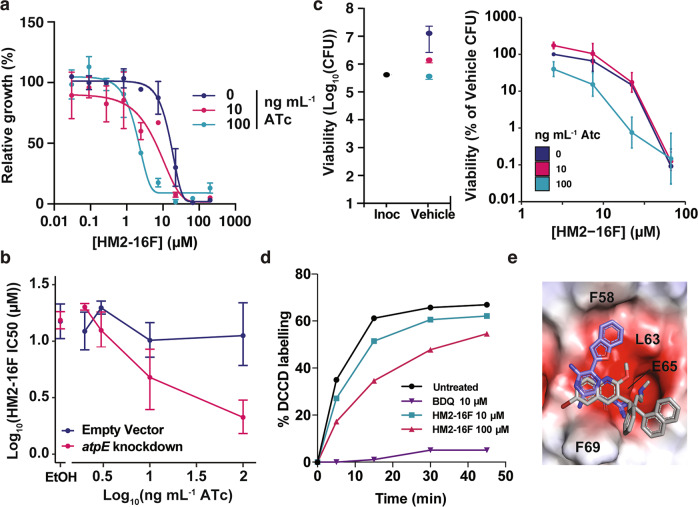
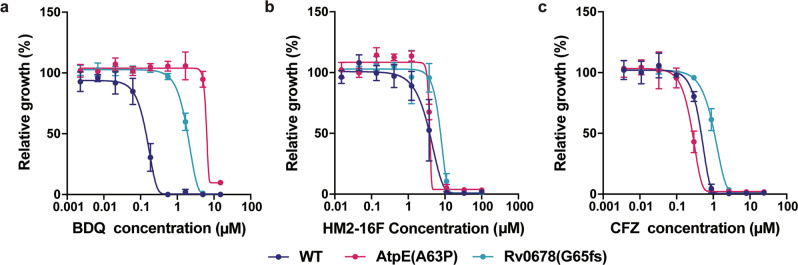
References
-
- World Health Organization. Global Tuberculosis Report 2019 (Geneva: World Health Organization, 2019).
-
- World Health Organization. Guidelines for the Treatment of Tuberculosis: Fourth Edition (World Health Organization, 2010).
-
- World Health Organization. Guidelines for the Programmatic Management of Drug-Resistant Tuberculosis: Emergency Update 2008 (Geneva: World Health Organization, 2008).
-
- Jones D. Tuberculosis success. Nat. Rev. Drug Discov. 2013;12:175–176. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
