

Deubiquitinase-targeting chimeras for targeted protein stabilization
- PMID: 35210618
- PMCID: PMC10125259
- DOI: 10.1038/s41589-022-00971-2
Deubiquitinase-targeting chimeras for targeted protein stabilization
Abstract
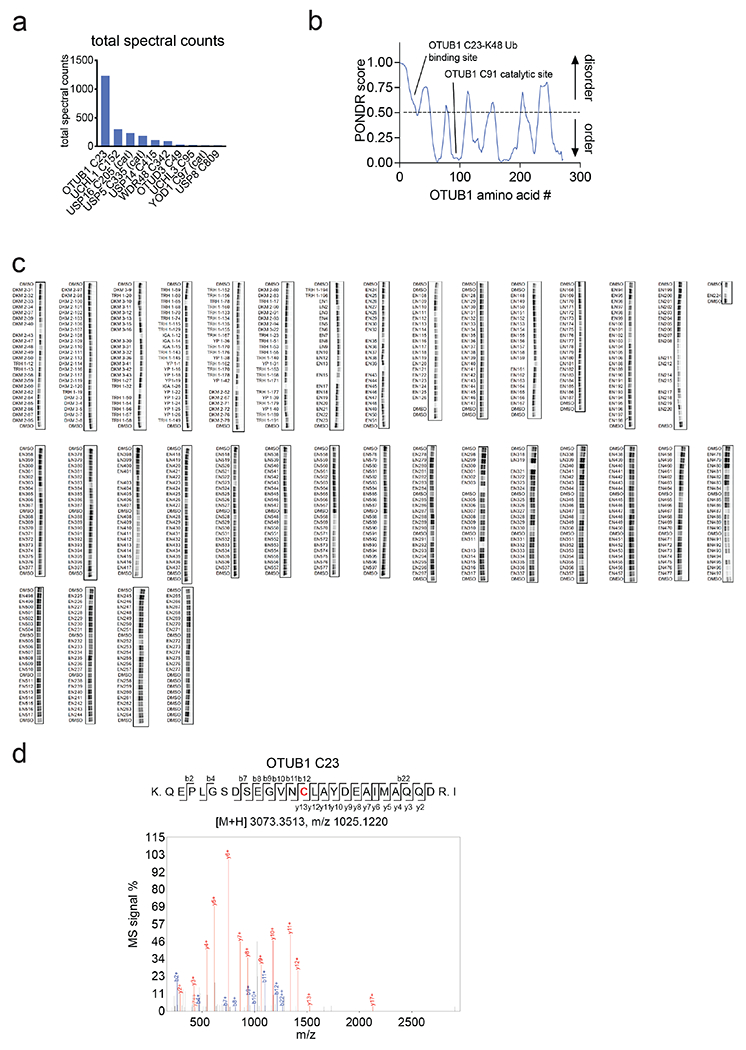
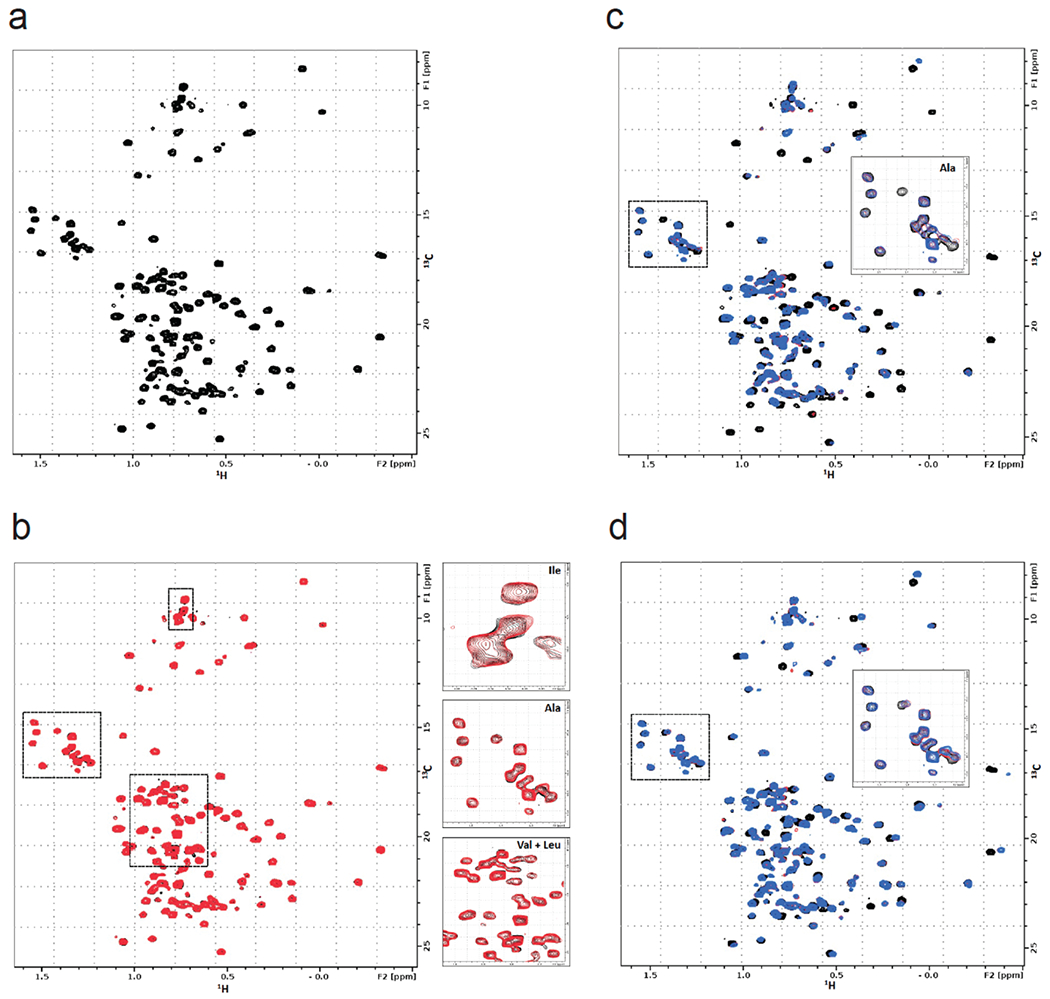
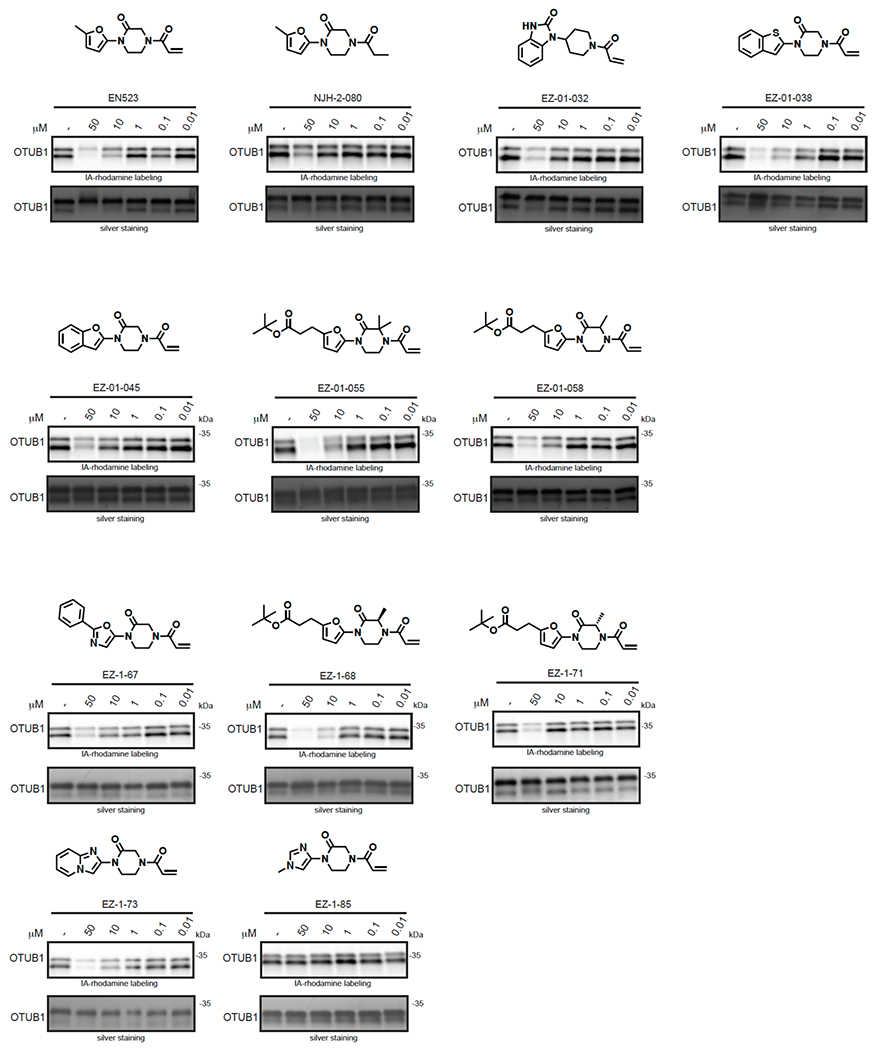
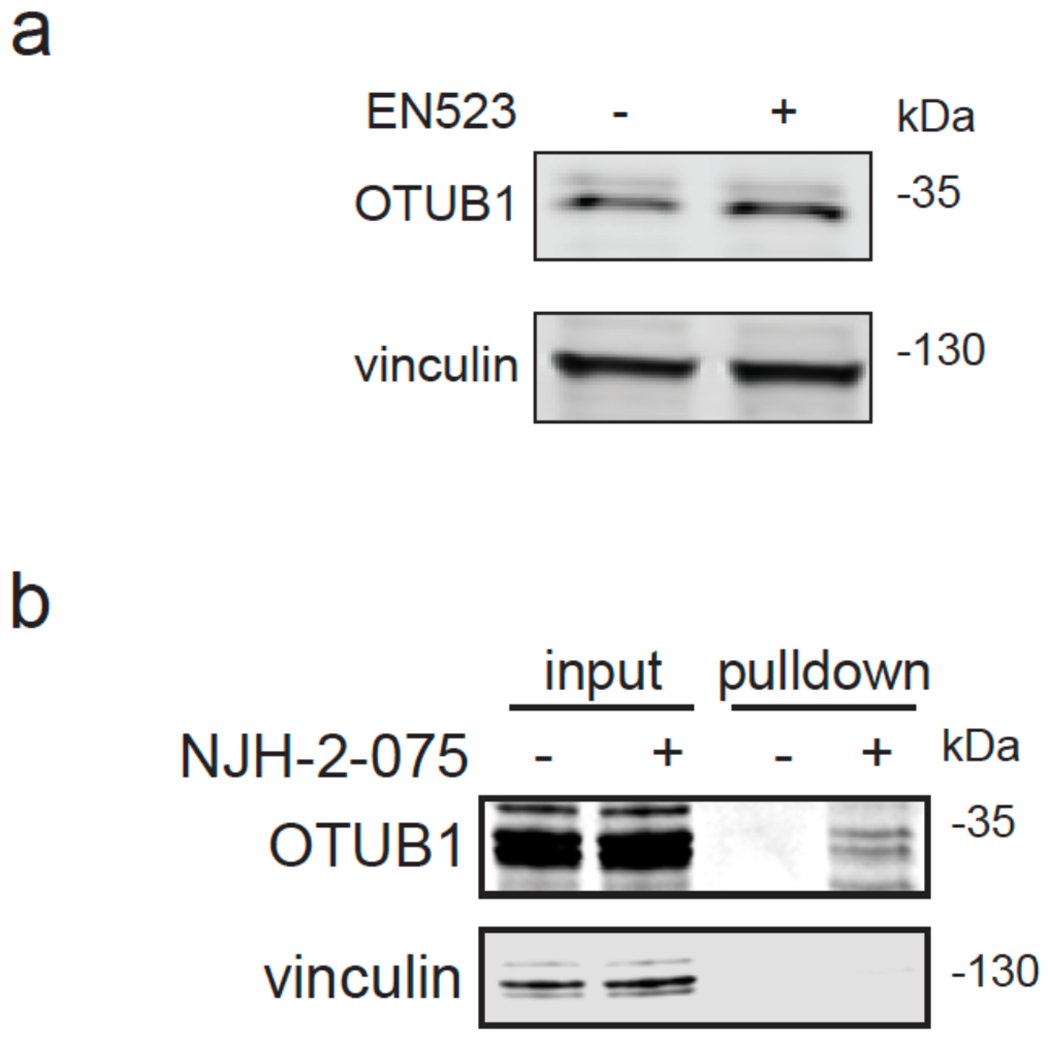
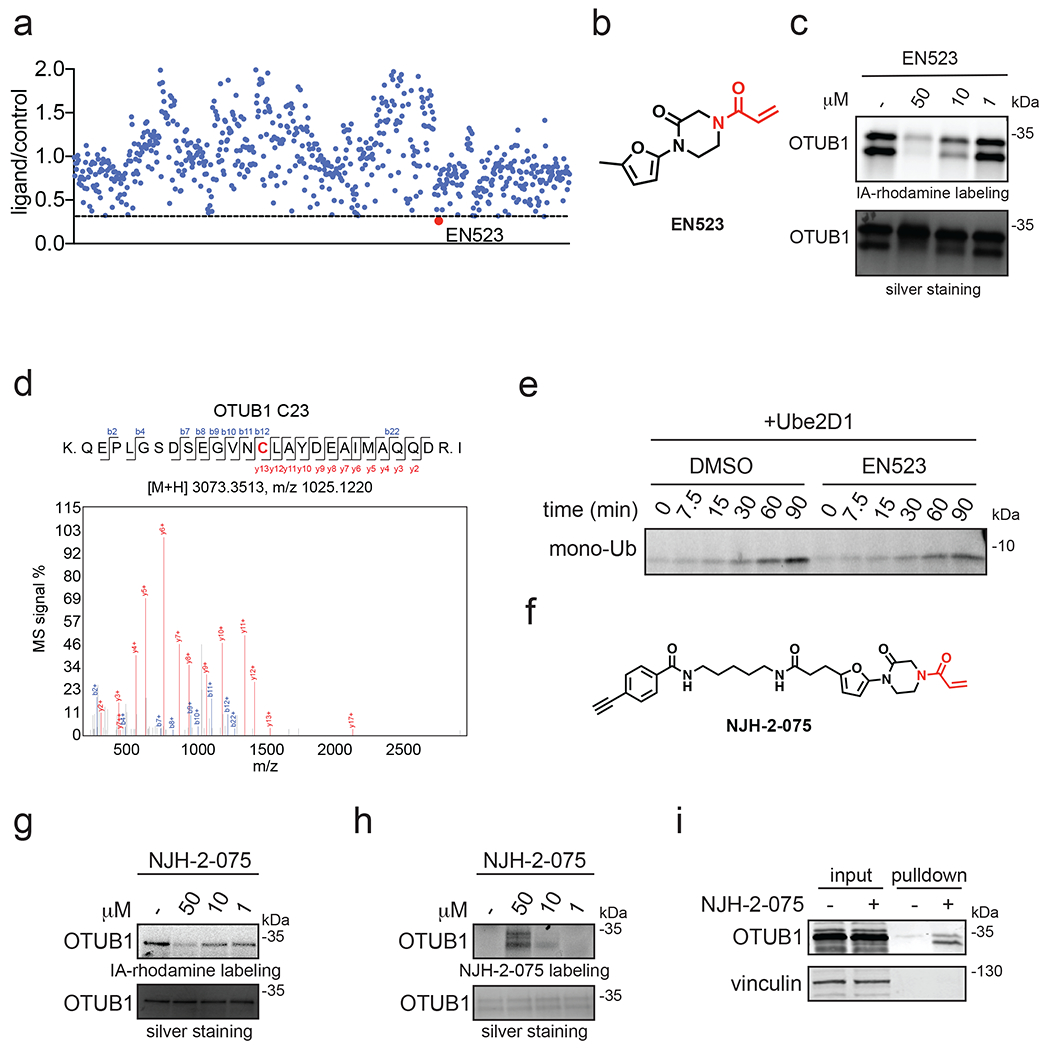
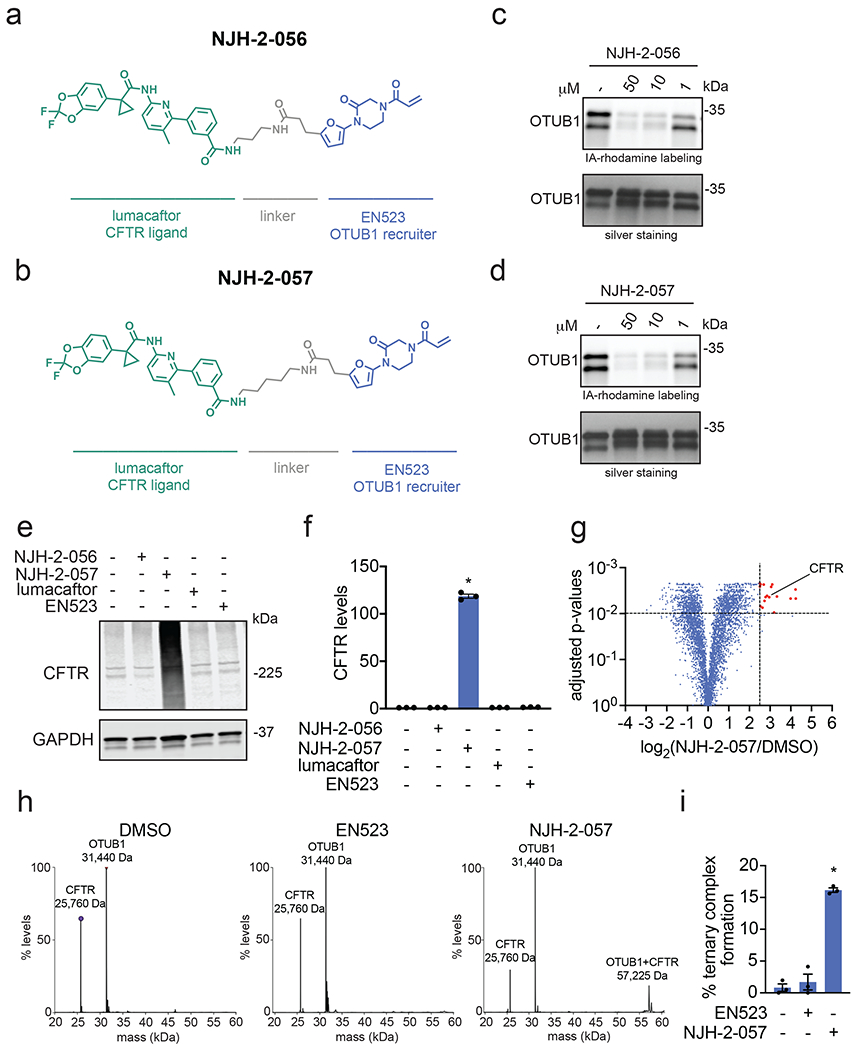
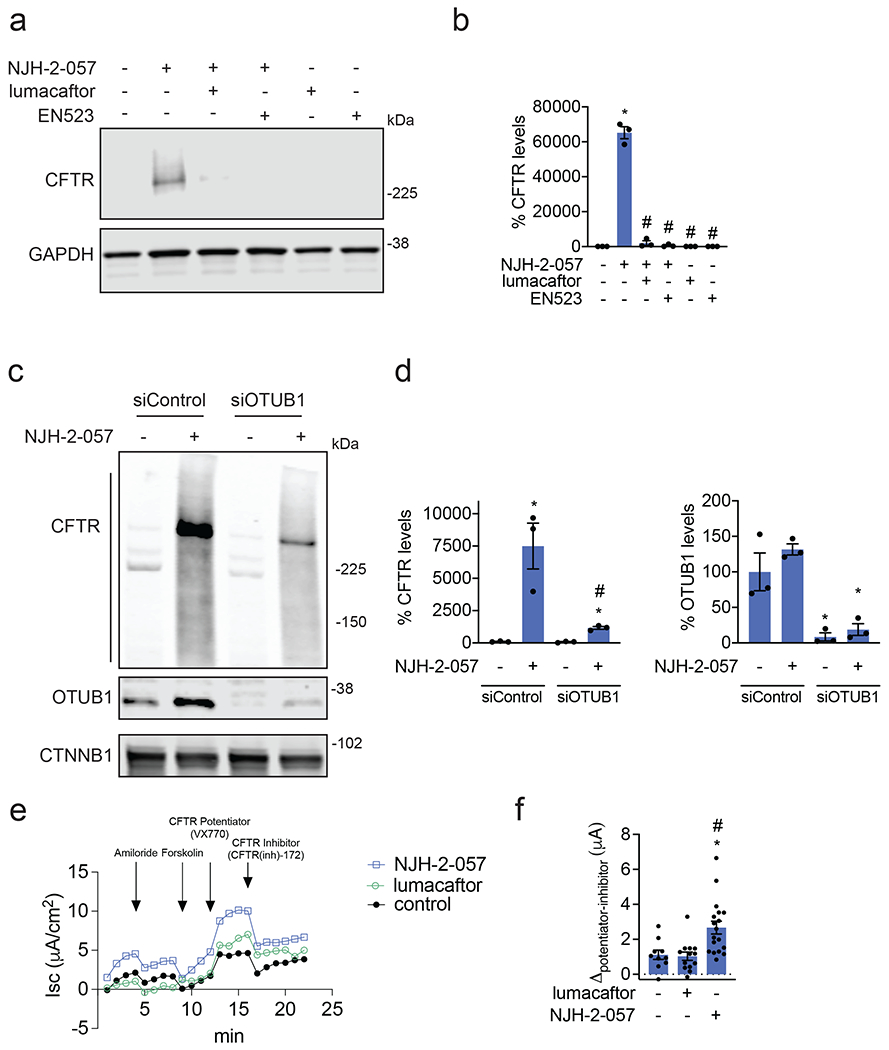
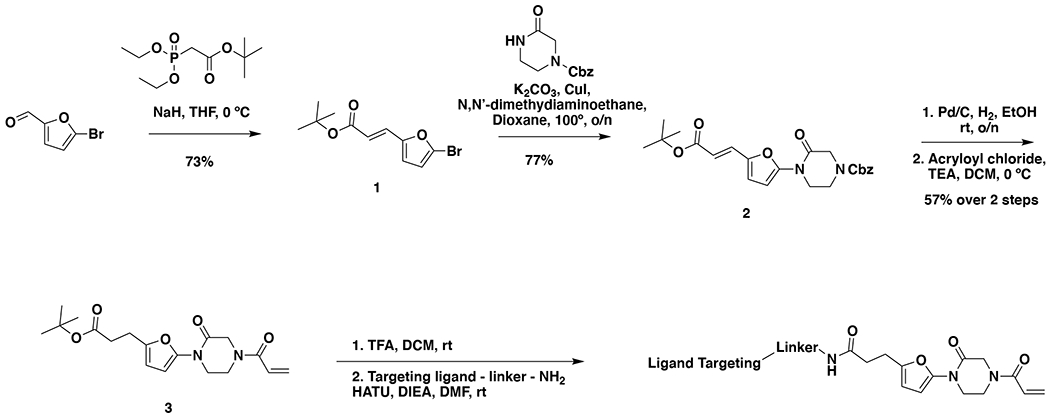
Many diseases are driven by proteins that are aberrantly ubiquitinated and degraded. These diseases would be therapeutically benefited by targeted protein stabilization (TPS). Here we present deubiquitinase-targeting chimeras (DUBTACs), heterobifunctional small molecules consisting of a deubiquitinase recruiter linked to a protein-targeting ligand, to stabilize the levels of specific proteins degraded in a ubiquitin-dependent manner. Using chemoproteomic approaches, we discovered the covalent ligand EN523 that targets a non-catalytic allosteric cysteine C23 in the K48-ubiquitin-specific deubiquitinase OTUB1. We showed that a DUBTAC consisting of our EN523 OTUB1 recruiter linked to lumacaftor, a drug used to treat cystic fibrosis that binds ΔF508-cystic fibrosis transmembrane conductance regulator (CFTR), robustly stabilized ΔF508-CFTR protein levels, leading to improved chloride channel conductance in human cystic fibrosis bronchial epithelial cells. We also demonstrated stabilization of the tumor suppressor kinase WEE1 in hepatoma cells. Our study showcases covalent chemoproteomic approaches to develop new induced proximity-based therapeutic modalities and introduces the DUBTAC platform for TPS.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures
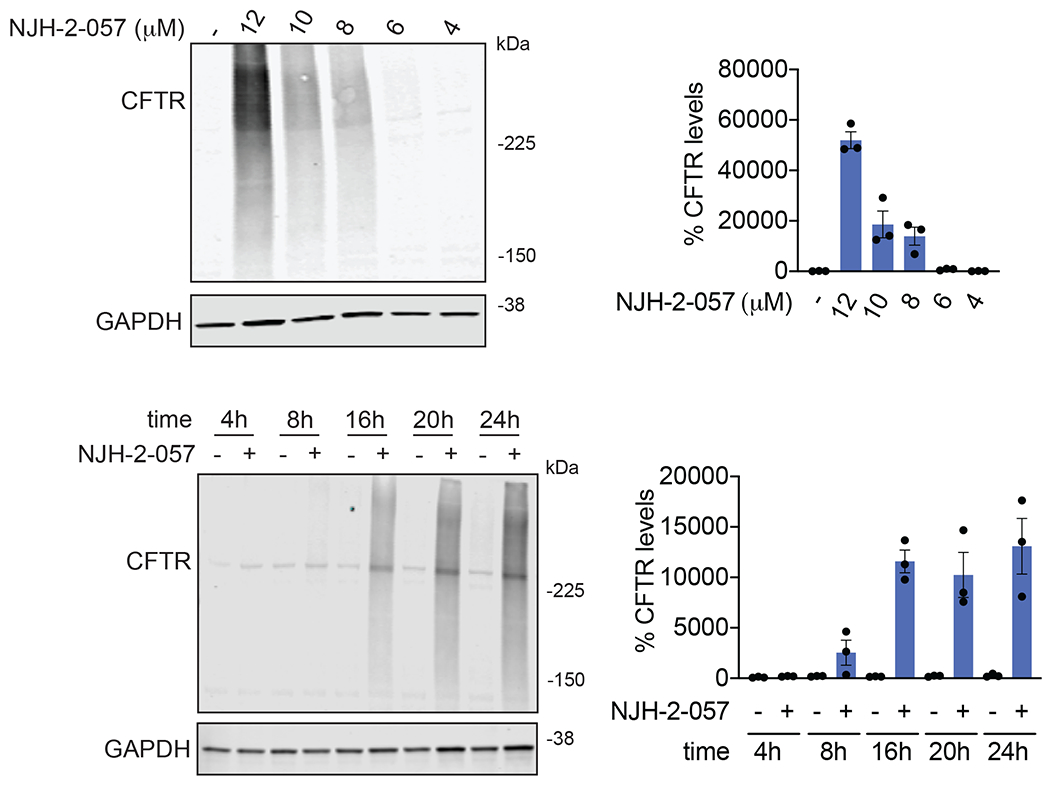
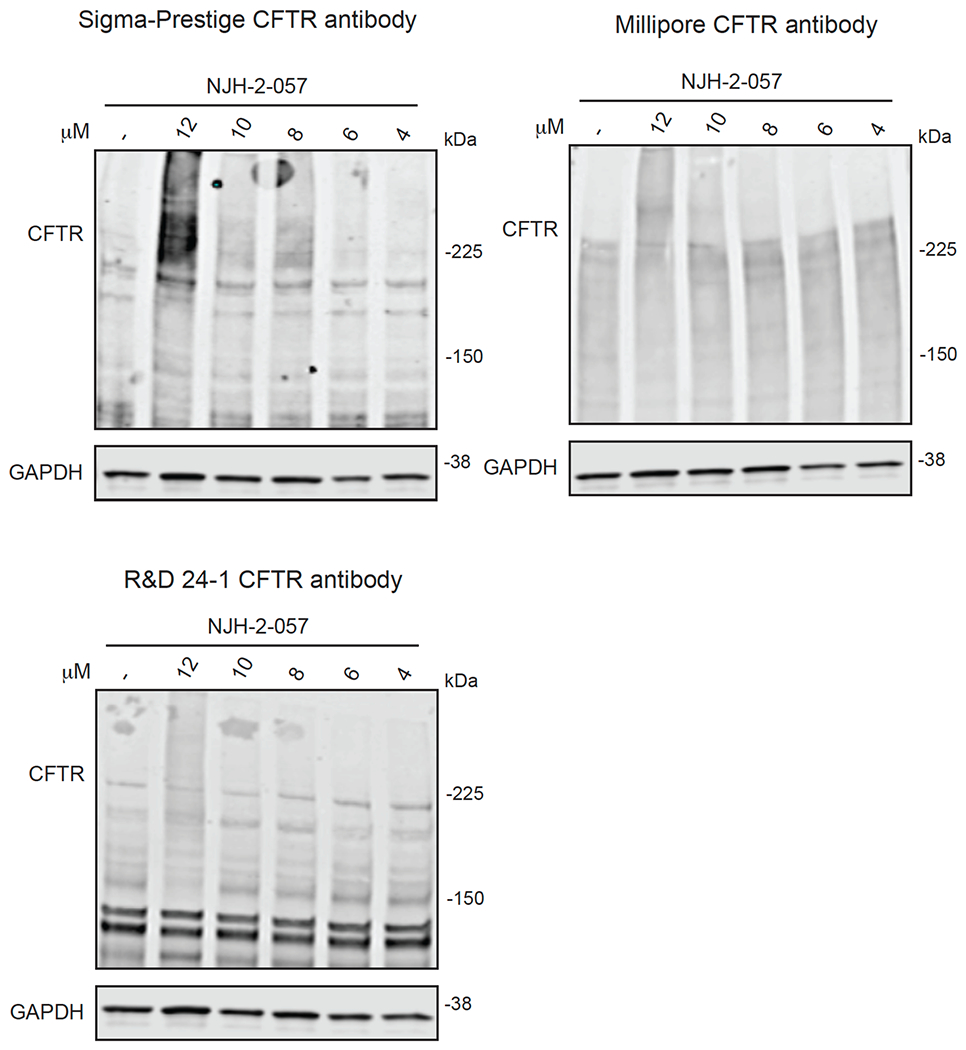
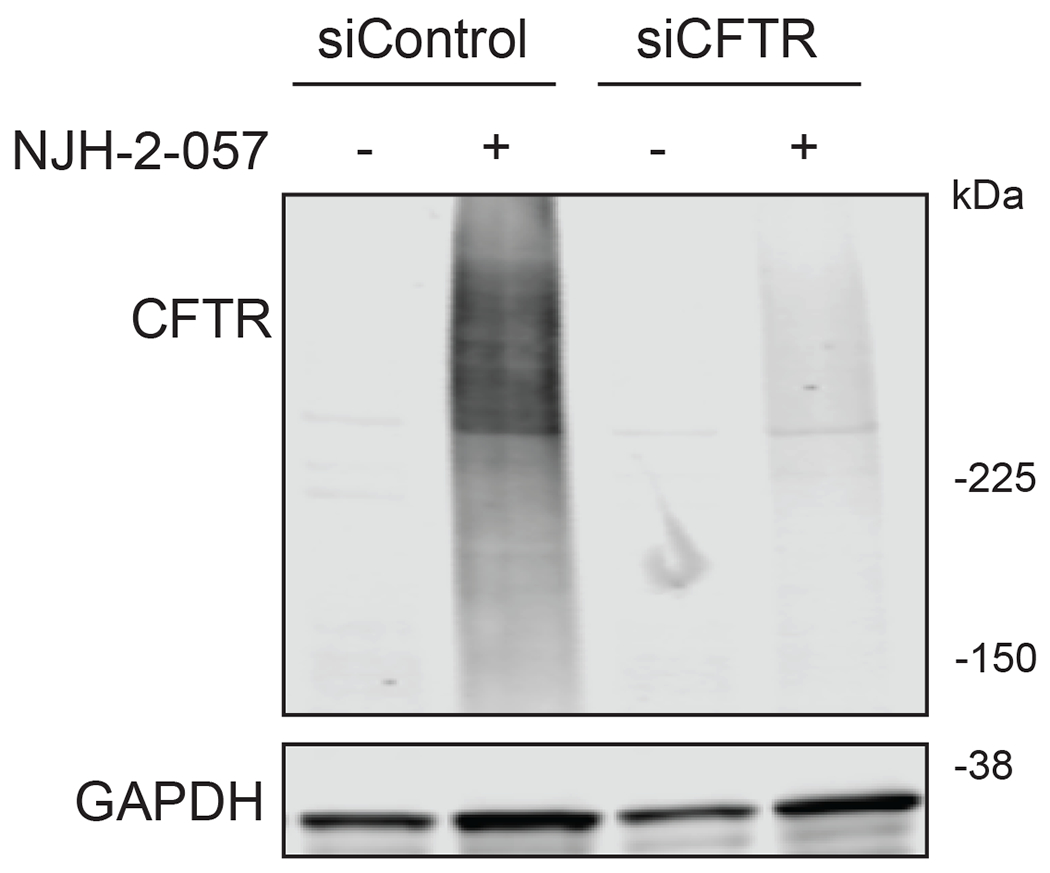
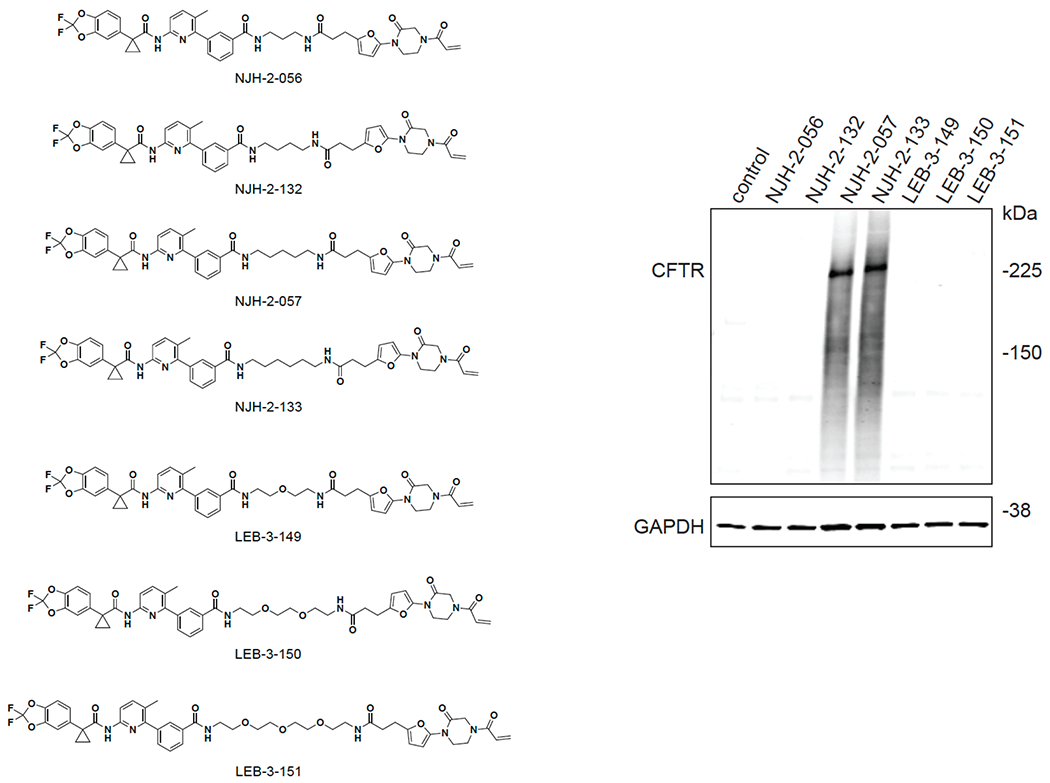
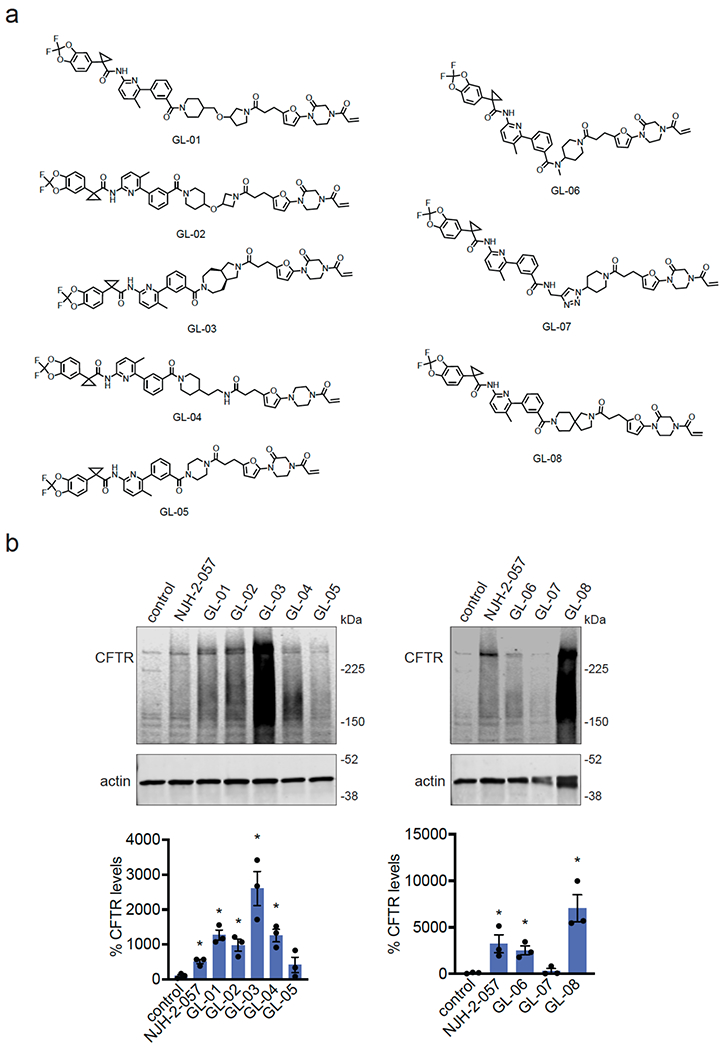
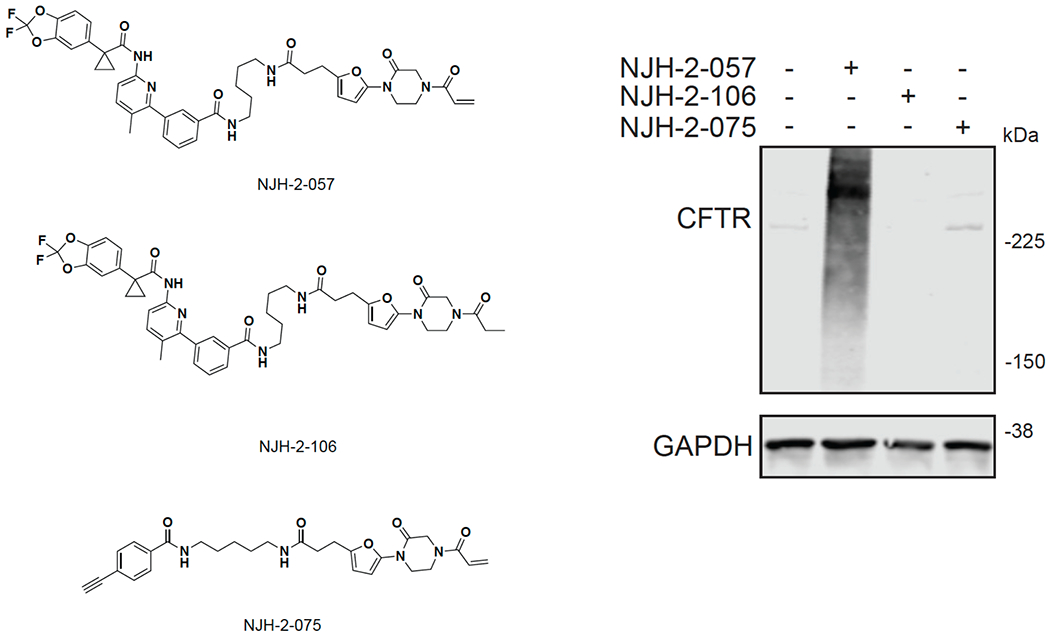
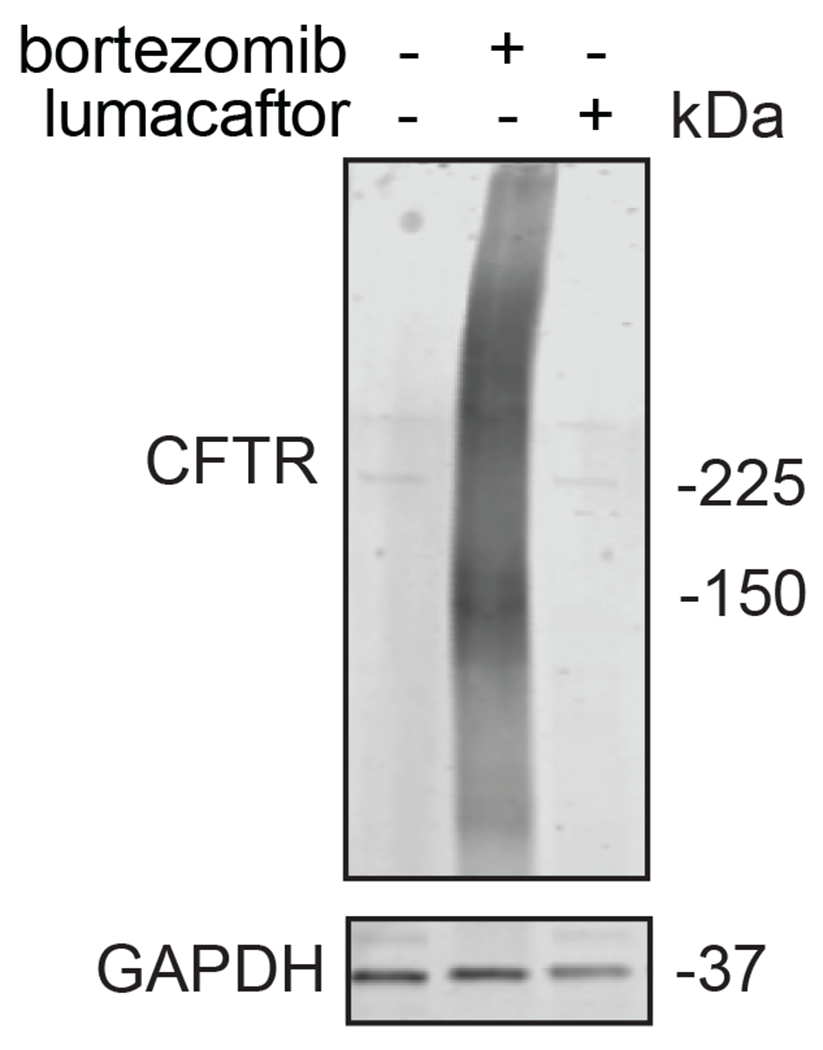
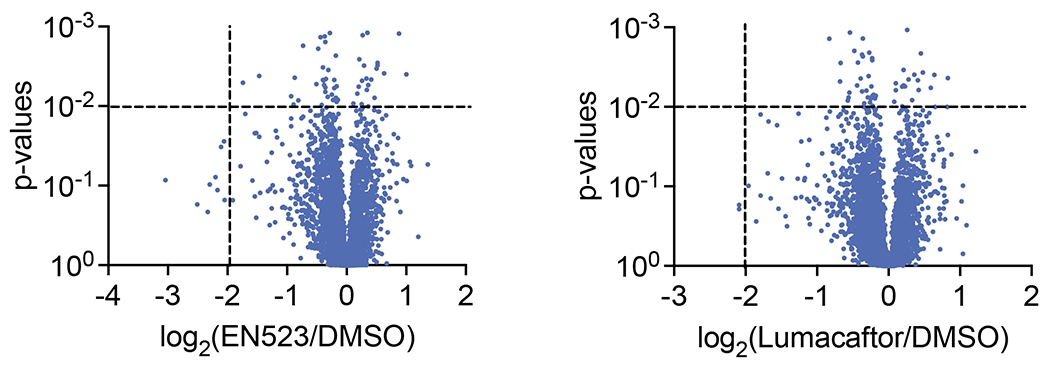
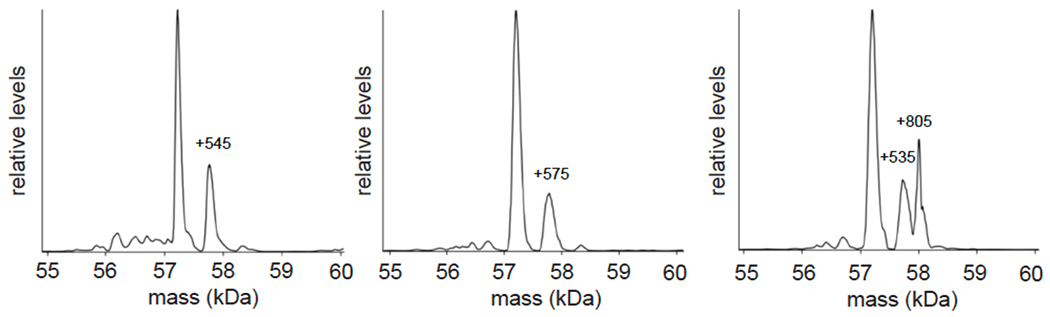
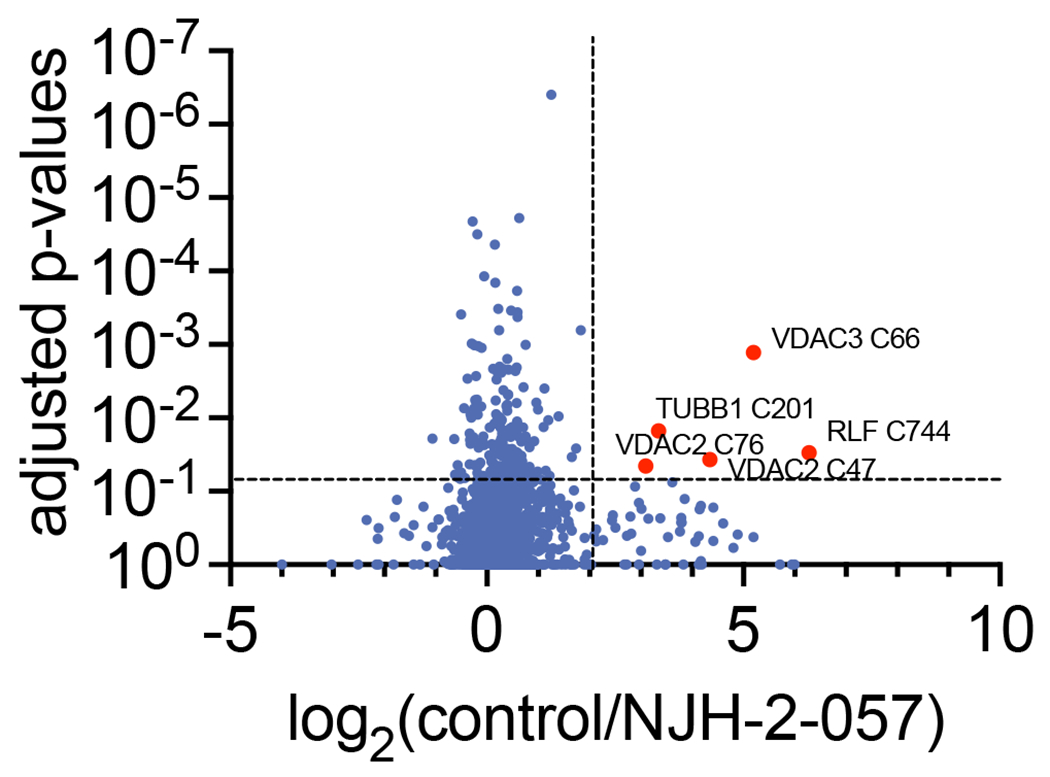
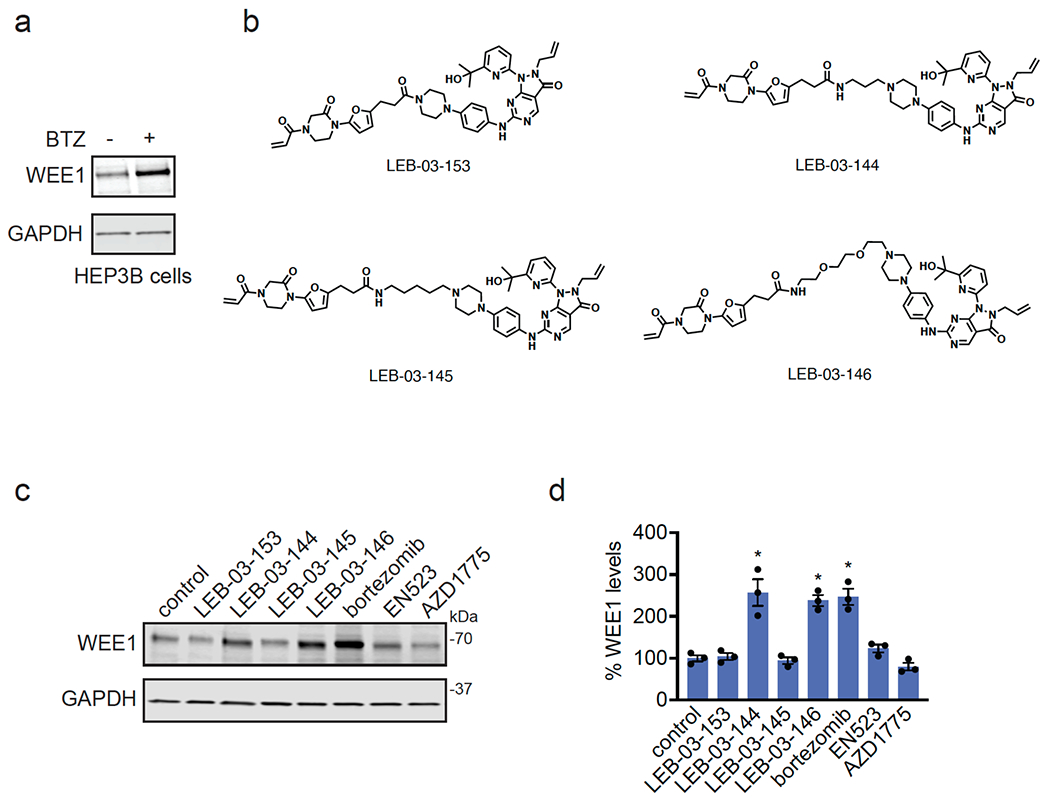
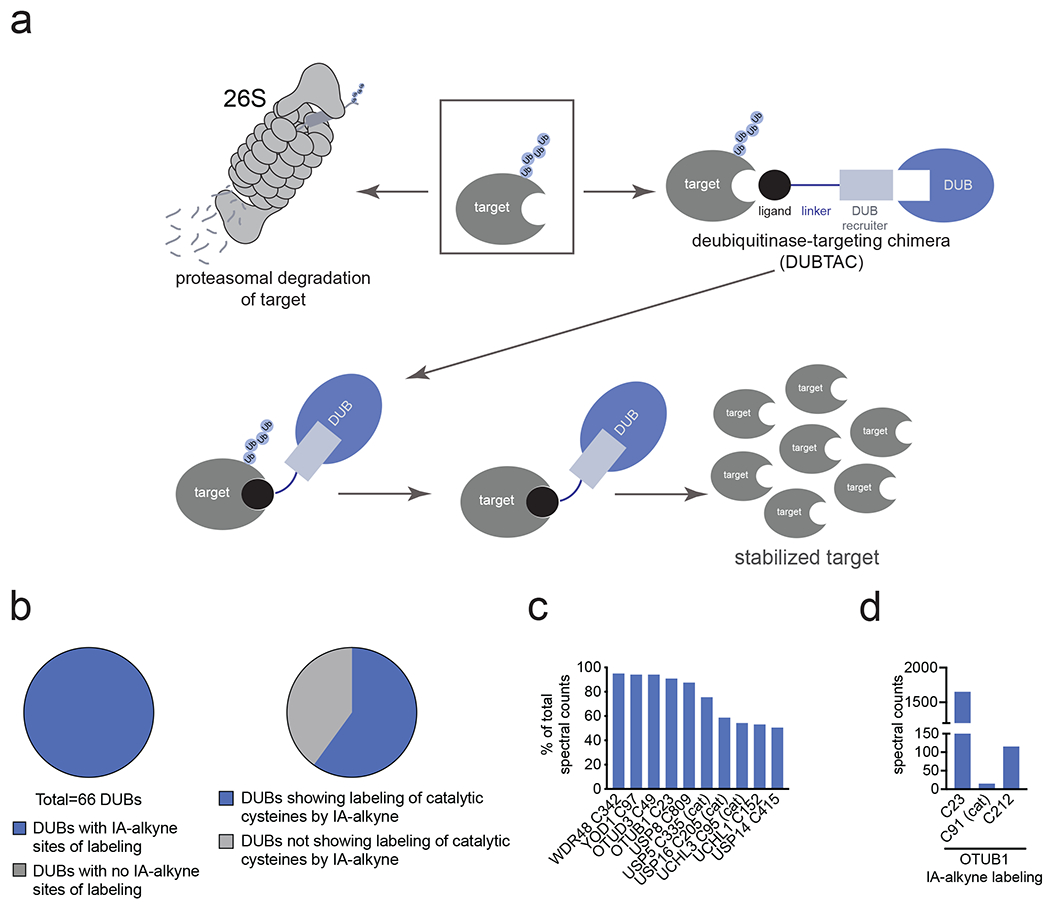
Comment in
-
DUB be good to me.Nat Chem Biol. 2022 Apr;18(4):358-359. doi: 10.1038/s41589-022-00978-9. Nat Chem Biol. 2022. PMID: 35210617 No abstract available.
-
DUBTACs for targeted protein stabilization.Nat Rev Drug Discov. 2022 Apr;21(4):258. doi: 10.1038/d41573-022-00039-9. Nat Rev Drug Discov. 2022. PMID: 35233100 No abstract available.
-
DUB to the rescue.Mol Cell. 2022 Apr 21;82(8):1411-1413. doi: 10.1016/j.molcel.2022.03.039. Mol Cell. 2022. PMID: 35452613
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
