

Advances in CAR-T Cell Genetic Engineering Strategies to Overcome Hurdles in Solid Tumors Treatment
- PMID: 35211124
- PMCID: PMC8861853
- DOI: 10.3389/fimmu.2022.830292
Advances in CAR-T Cell Genetic Engineering Strategies to Overcome Hurdles in Solid Tumors Treatment
Abstract
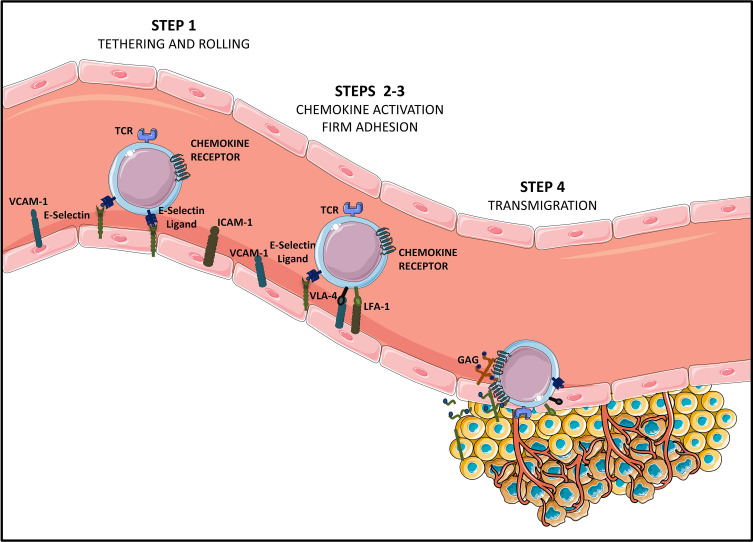
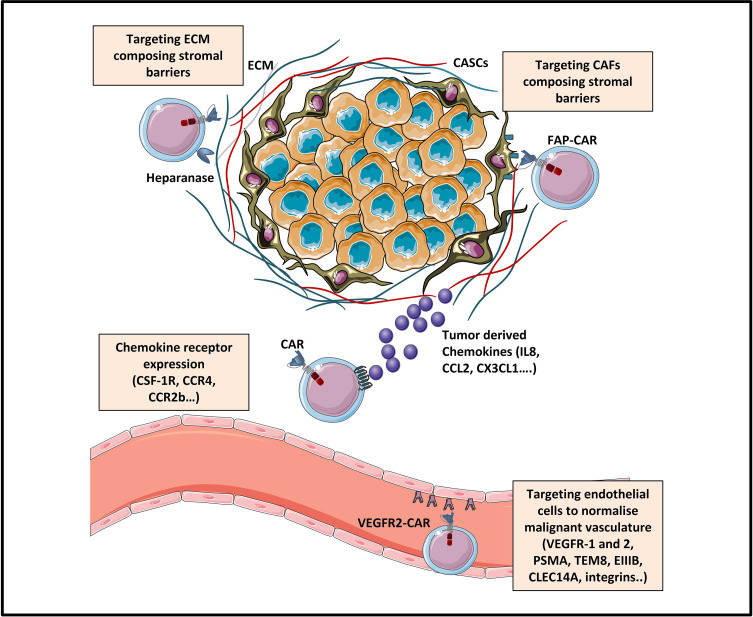
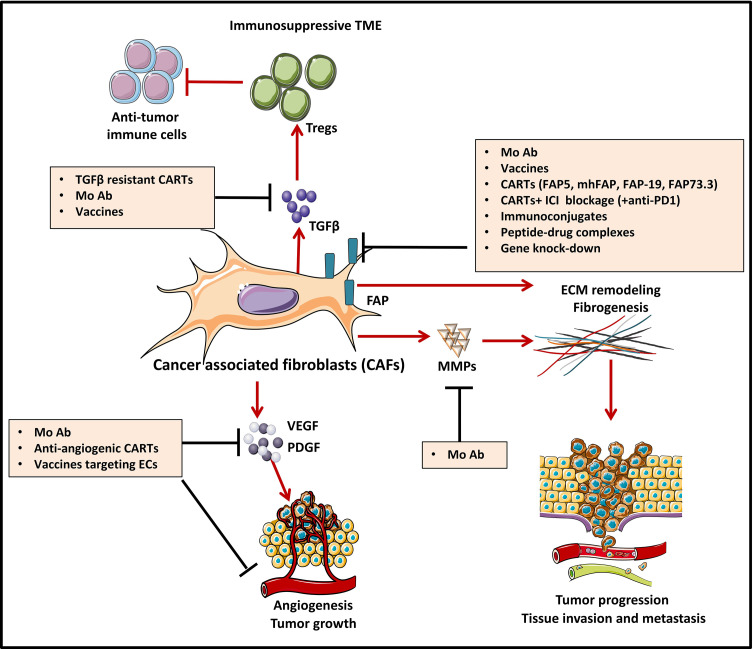
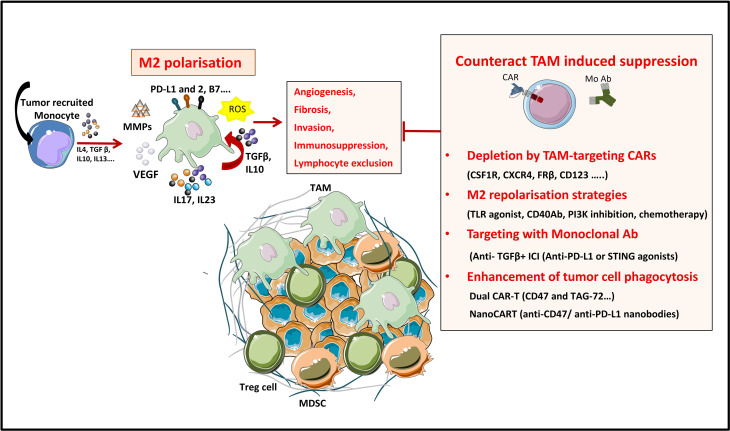
During this last decade, adoptive transfer of T lymphocytes genetically modified to express chimeric antigen receptors (CARs) emerged as a valuable therapeutic strategy in hematological cancers. However, this immunotherapy has demonstrated limited efficacy in solid tumors. The main obstacle encountered by CAR-T cells in solid malignancies is the immunosuppressive tumor microenvironment (TME). The TME impedes tumor trafficking and penetration of T lymphocytes and installs an immunosuppressive milieu by producing suppressive soluble factors and by overexpressing negative immune checkpoints. In order to overcome these hurdles, new CAR-T cells engineering strategies were designed, to potentiate tumor recognition and infiltration and anti-cancer activity in the hostile TME. In this review, we provide an overview of the major mechanisms used by tumor cells to evade immune defenses and we critically expose the most optimistic engineering strategies to make CAR-T cell therapy a solid option for solid tumors.
Keywords: Angiogenesis; CAR-T cell immunotherapy; Chemokines; Immune checkpoints; Solid tumor; Tumor Homing; Tumor microenvironment; Tumor stroma.
Copyright © 2022 Andrea, Chiron, Mallah, Bessoles, Sarrabayrouse and Hacein-Bey-Abina.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Manipulating the tumor microenvironment by adoptive cell transfer of CAR T-cells.Mamm Genome. 2018 Dec;29(11-12):739-756. doi: 10.1007/s00335-018-9756-5. Epub 2018 Jul 9. Mamm Genome. 2018. PMID: 29987406 Review.
-
Solid Tumors Challenges and New Insights of CAR T Cell Engineering.Stem Cell Rev Rep. 2019 Oct;15(5):619-636. doi: 10.1007/s12015-019-09901-7. Stem Cell Rev Rep. 2019. PMID: 31161552 Review.
-
Engineering CAR-T Cells for Next-Generation Cancer Therapy.Cancer Cell. 2020 Oct 12;38(4):473-488. doi: 10.1016/j.ccell.2020.07.005. Epub 2020 Jul 30. Cancer Cell. 2020. PMID: 32735779 Review.
-
Chimeric antigen receptor-engineered T-cell therapy for liver cancer.Hepatobiliary Pancreat Dis Int. 2018 Aug;17(4):301-309. doi: 10.1016/j.hbpd.2018.05.005. Epub 2018 May 24. Hepatobiliary Pancreat Dis Int. 2018. PMID: 29861325 Review.
-
Perspectives on Chimeric Antigen Receptor T-Cell Immunotherapy for Solid Tumors.Front Immunol. 2018 May 22;9:1104. doi: 10.3389/fimmu.2018.01104. eCollection 2018. Front Immunol. 2018. PMID: 29872437 Free PMC article. Review.
Cited by
-
Advanced Strategies of CAR-T Cell Therapy in Solid Tumors and Hematological Malignancies.Recent Pat Anticancer Drug Discov. 2024;19(5):557-572. doi: 10.2174/0115748928277331231218115402. Recent Pat Anticancer Drug Discov. 2024. PMID: 38213150 Review.
-
Immunotherapy Innovations in the Fight against Osteosarcoma: Emerging Strategies and Promising Progress.Pharmaceutics. 2024 Feb 8;16(2):251. doi: 10.3390/pharmaceutics16020251. Pharmaceutics. 2024. PMID: 38399305 Free PMC article. Review.
-
Advances in CAR T Cell Therapy for Non-Small Cell Lung Cancer.Curr Issues Mol Biol. 2023 Nov 12;45(11):9019-9038. doi: 10.3390/cimb45110566. Curr Issues Mol Biol. 2023. PMID: 37998743 Free PMC article. Review.
-
Molecular and therapeutic effect of CRISPR in treating cancer.Med Oncol. 2023 Jan 17;40(2):81. doi: 10.1007/s12032-022-01930-6. Med Oncol. 2023. Retraction in: Med Oncol. 2025 Mar 24;42(5):132. doi: 10.1007/s12032-025-02692-7. PMID: 36650384 Free PMC article. Retracted. Review.
-
Recent Advances and Challenges in Cancer Immunotherapy.Cancers (Basel). 2022 Aug 17;14(16):3972. doi: 10.3390/cancers14163972. Cancers (Basel). 2022. PMID: 36010965 Free PMC article. Review.
References
-
- Chmielewski M, Abken H. TRUCKS, the Fourth-Generation CAR T Cells: Current Developments and Clinical Translation. Adv IN Cell AND Gene Ther (2020) 3:e84. doi: 10.1002/acg2.84 - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical