

Signature-scoring methods developed for bulk samples are not adequate for cancer single-cell RNA sequencing data
- PMID: 35212622
- PMCID: PMC8916770
- DOI: 10.7554/eLife.71994
Signature-scoring methods developed for bulk samples are not adequate for cancer single-cell RNA sequencing data
Abstract
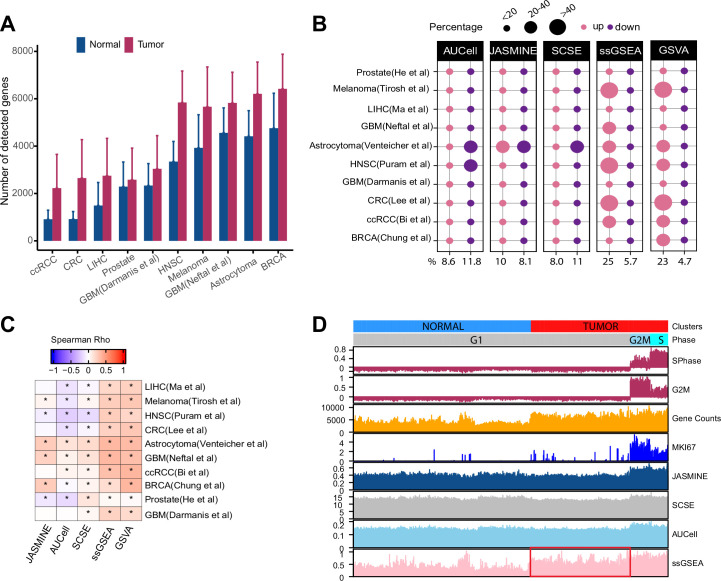
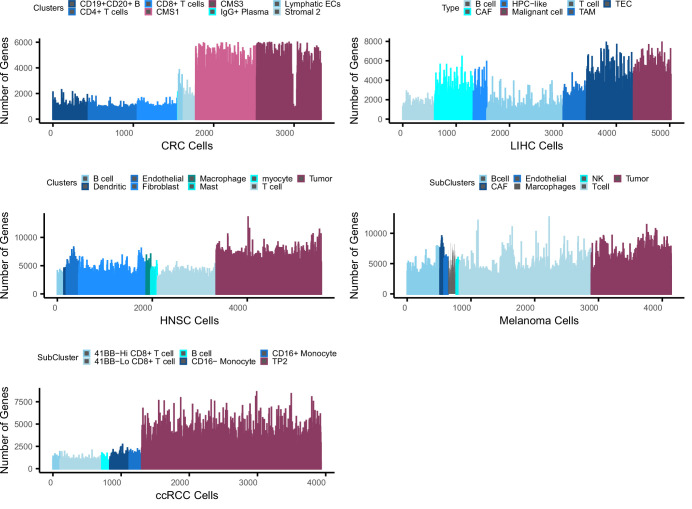
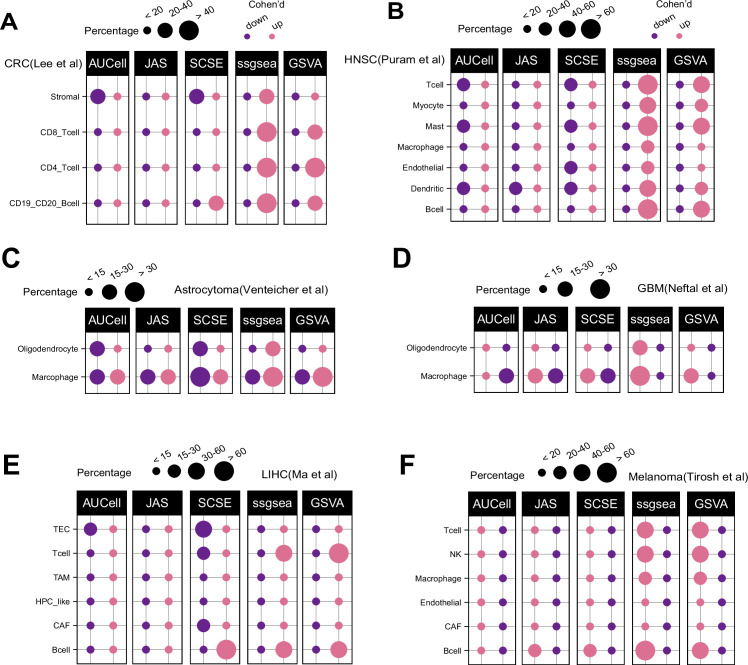
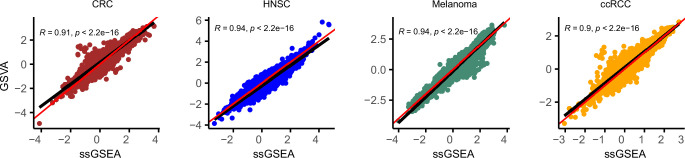
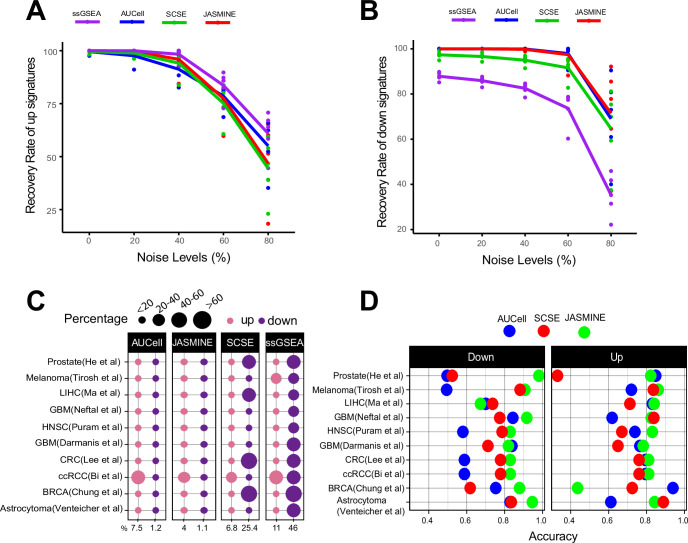
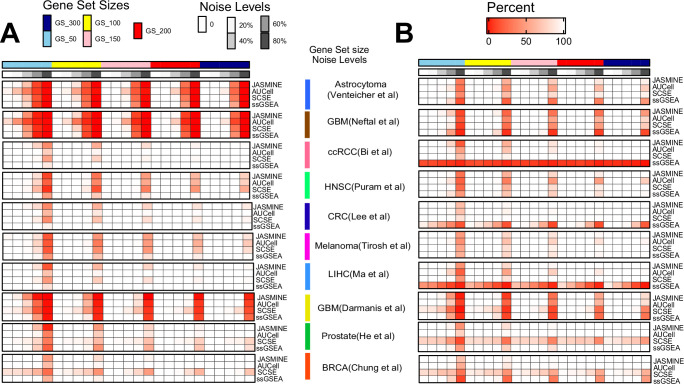
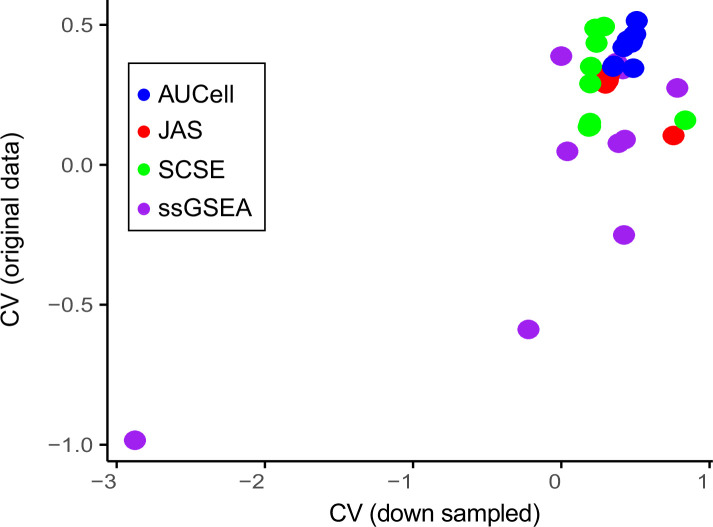
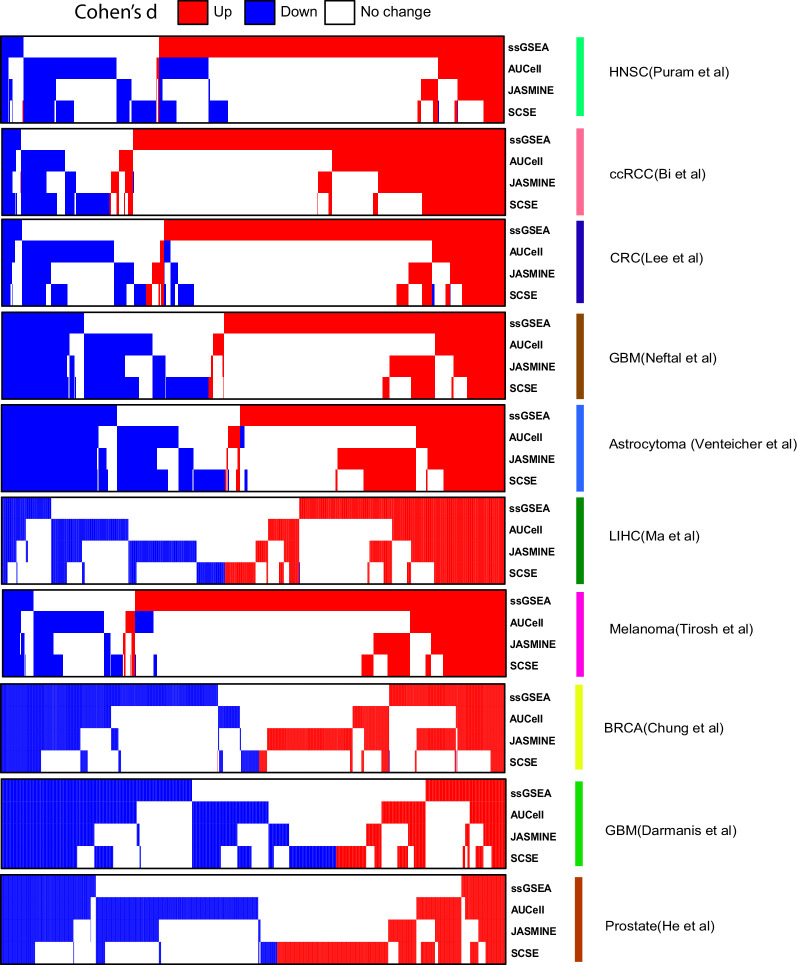
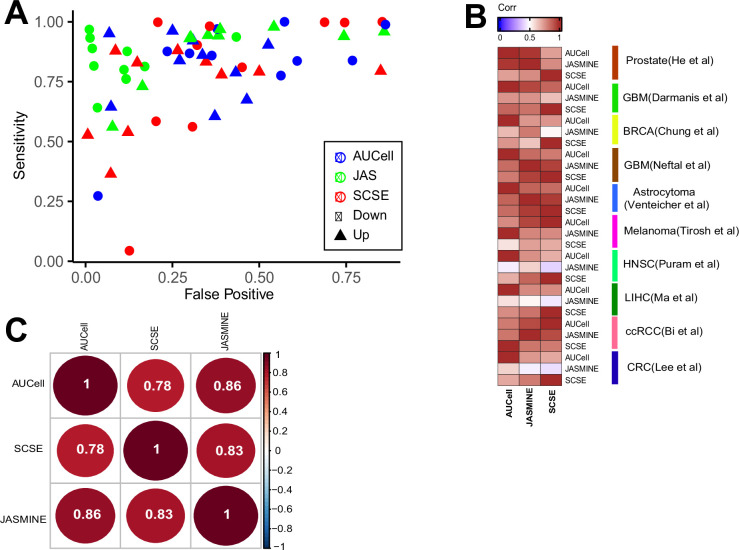
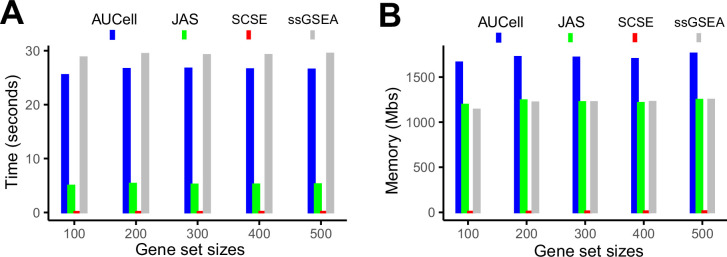
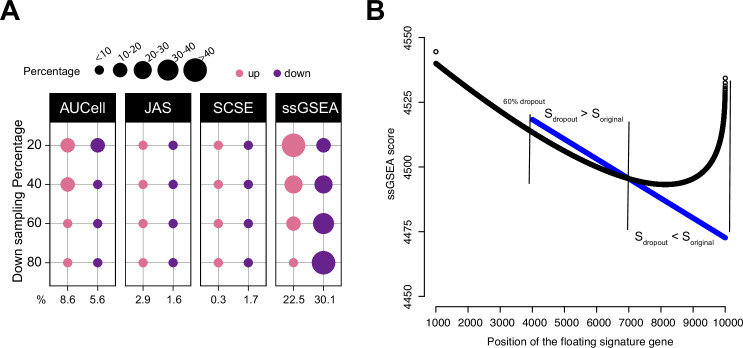
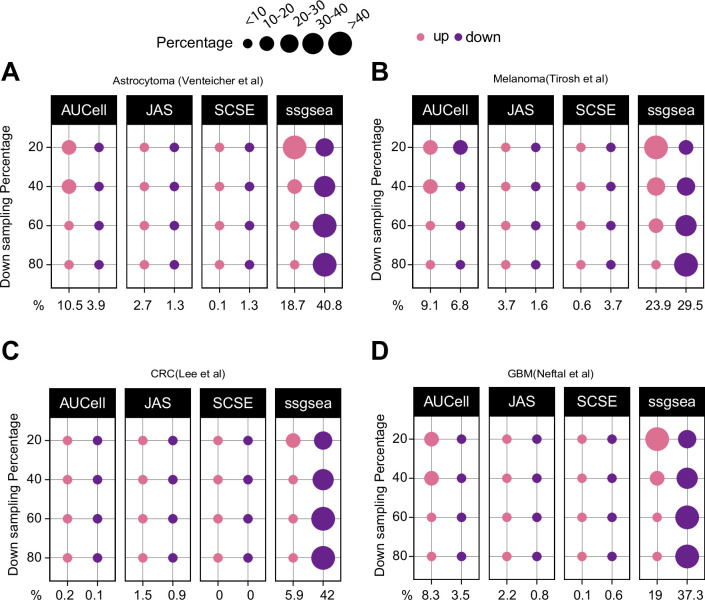
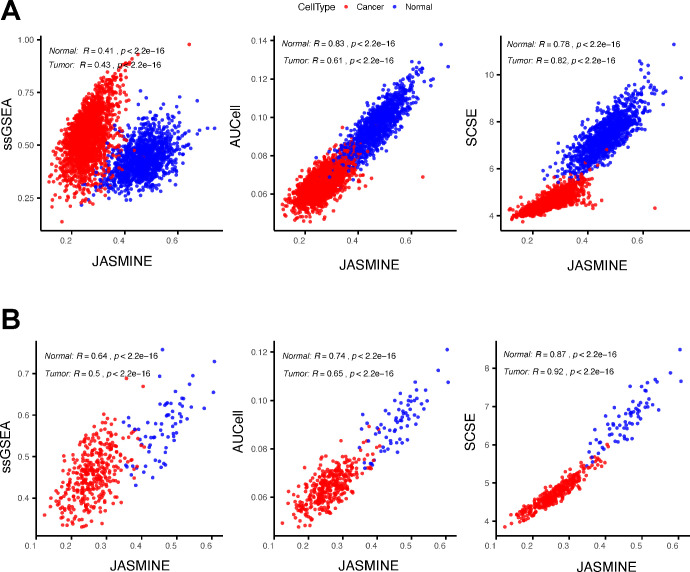
Quantifying the activity of gene expression signatures is common in analyses of single-cell RNA sequencing data. Methods originally developed for bulk samples are often used for this purpose without accounting for contextual differences between bulk and single-cell data. More broadly, few attempts have been made to benchmark these methods. Here, we benchmark five such methods, including single sample gene set enrichment analysis (ssGSEA), Gene Set Variation Analysis (GSVA), AUCell, Single Cell Signature Explorer (SCSE), and a new method we developed, Jointly Assessing Signature Mean and Inferring Enrichment (JASMINE). Using cancer as an example, we show cancer cells consistently express more genes than normal cells. This imbalance leads to bias in performance by bulk-sample-based ssGSEA in gold standard tests and down sampling experiments. In contrast, single-cell-based methods are less susceptible. Our results suggest caution should be exercised when using bulk-sample-based methods in single-cell data analyses, and cellular contexts should be taken into consideration when designing benchmarking strategies.
Keywords: benchmarking; cancer biology; cancer stemness; computational biology; gene counts; human; signature scoring; single cell RNA sequencing; systems biology.
© 2022, Noureen et al.
Conflict of interest statement
NN, ZY, YC, XW, SZ No competing interests declared
Figures
References
-
- Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine J-C, Geurts P, Aerts J, van den Oord J, Atak ZK, Wouters J, Aerts S. SCENIC: single-cell regulatory network inference and clustering. Nature Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. - DOI - PMC - PubMed
-
- Ben-Shachar M, Lüdecke D, Makowski D. effectsize: Estimation of Effect Size Indices and Standardized Parameters. Journal of Open Source Software. 2020;5:2815. doi: 10.21105/joss.02815. - DOI
-
- Bi K, He MX, Bakouny Z, Kanodia A, Napolitano S, Wu J, Grimaldi G, Braun DA, Cuoco MS, Mayorga A, DelloStritto L, Bouchard G, Steinharter J, Tewari AK, Vokes NI, Shannon E, Sun M, Park J, Chang SL, McGregor BA, Haq R, Denize T, Signoretti S, Guerriero JL, Vigneau S, Rozenblatt-Rosen O, Rotem A, Regev A, Choueiri TK, Van Allen EM. Tumor and immune reprogramming during immunotherapy in advanced renal cell carcinoma. Cancer Cell. 2021;39:649–661. doi: 10.1016/j.ccell.2021.02.015. - DOI - PMC - PubMed
Publication types
MeSH terms
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
