Single-cell eQTL analysis of activated T cell subsets reveals activation and cell type-dependent effects of disease-risk variants
- PMID: 35213211
- PMCID: PMC9035271
- DOI: 10.1126/sciimmunol.abm2508
Single-cell eQTL analysis of activated T cell subsets reveals activation and cell type-dependent effects of disease-risk variants
Abstract
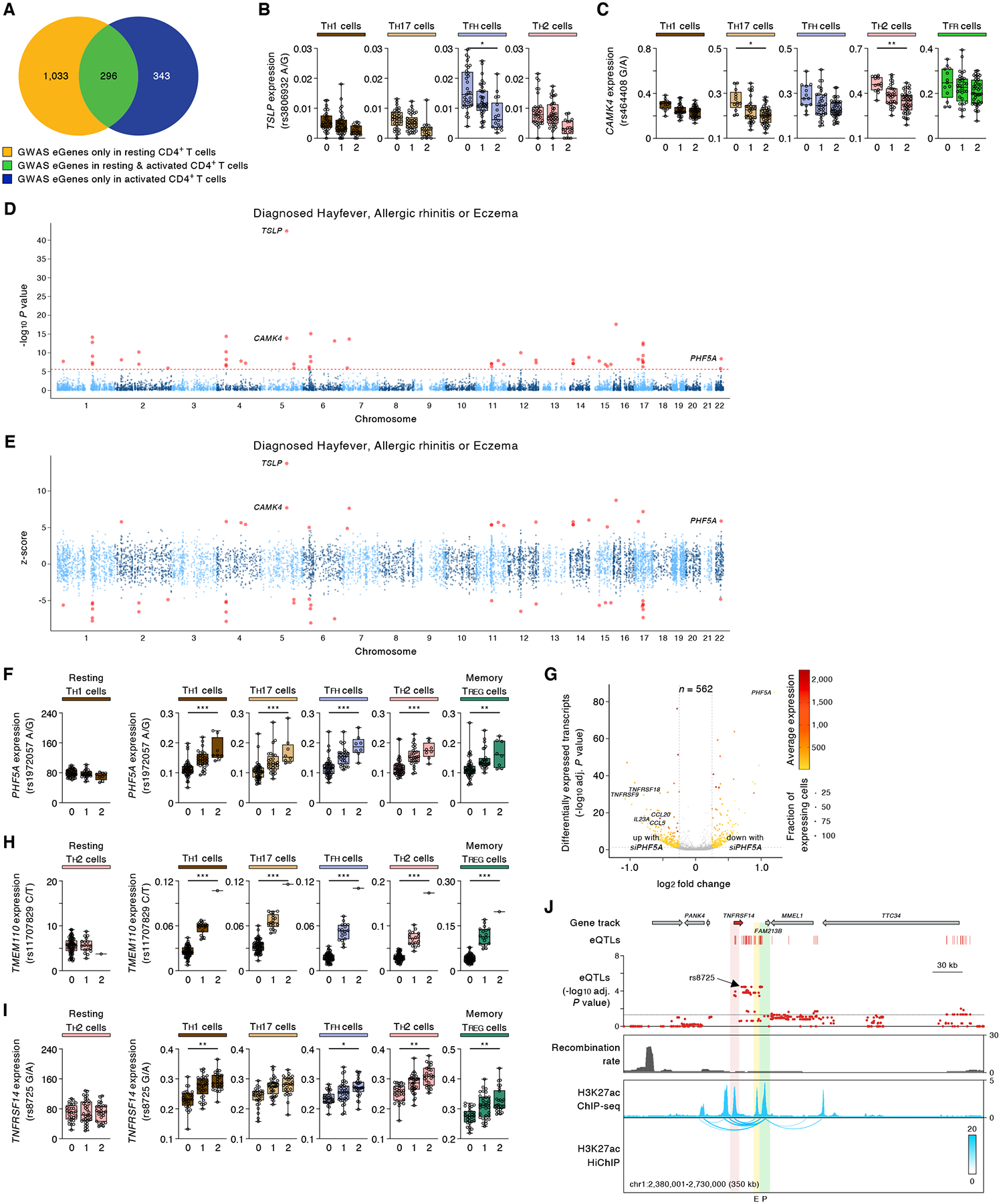
The impact of genetic variants on cells challenged in biologically relevant contexts has not been fully explored. Here, we activated CD4+ T cells from 89 healthy donors and performed a single-cell RNA sequencing assay with >1 million cells to examine cell type-specific and activation-dependent effects of genetic variants. Single-cell expression quantitative trait loci (sc-eQTL) analysis of 19 distinct CD4+ T cell subsets showed that the expression of over 4000 genes is significantly associated with common genetic polymorphisms and that most of these genes show their most prominent effects in specific cell types. These genes included many that encode for molecules important for activation, differentiation, and effector functions of T cells. We also found new gene associations for disease-risk variants identified from genome-wide association studies and highlighted the cell types in which their effects are most prominent. We found that biological sex has a major influence on activation-dependent gene expression in CD4+ T cell subsets. Sex-biased transcripts were significantly enriched in several pathways that are essential for the initiation and execution of effector functions by CD4+ T cells like TCR signaling, cytokines, cytokine receptors, costimulatory, apoptosis, and cell-cell adhesion pathways. Overall, this DICE (Database of Immune Cell Expression, eQTLs, and Epigenomics) subproject highlights the power of sc-eQTL studies for simultaneously exploring the activation and cell type-dependent effects of common genetic variants on gene expression (https://dice-database.org).
Conflict of interest statement
Competing interests
The authors declare no competing financial interests.
Figures




References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials