

Genome-Wide Analysis and Characterization of the Proline-Rich Extensin-like Receptor Kinases (PERKs) Gene Family Reveals Their Role in Different Developmental Stages and Stress Conditions in Wheat (Triticum aestivum L.)
- PMID: 35214830
- PMCID: PMC8880425
- DOI: 10.3390/plants11040496
Genome-Wide Analysis and Characterization of the Proline-Rich Extensin-like Receptor Kinases (PERKs) Gene Family Reveals Their Role in Different Developmental Stages and Stress Conditions in Wheat (Triticum aestivum L.)
Abstract
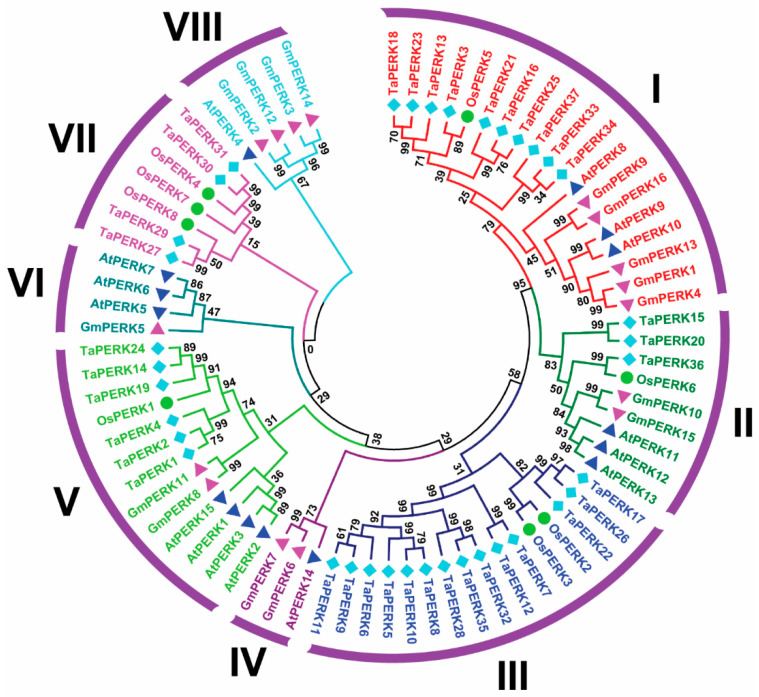
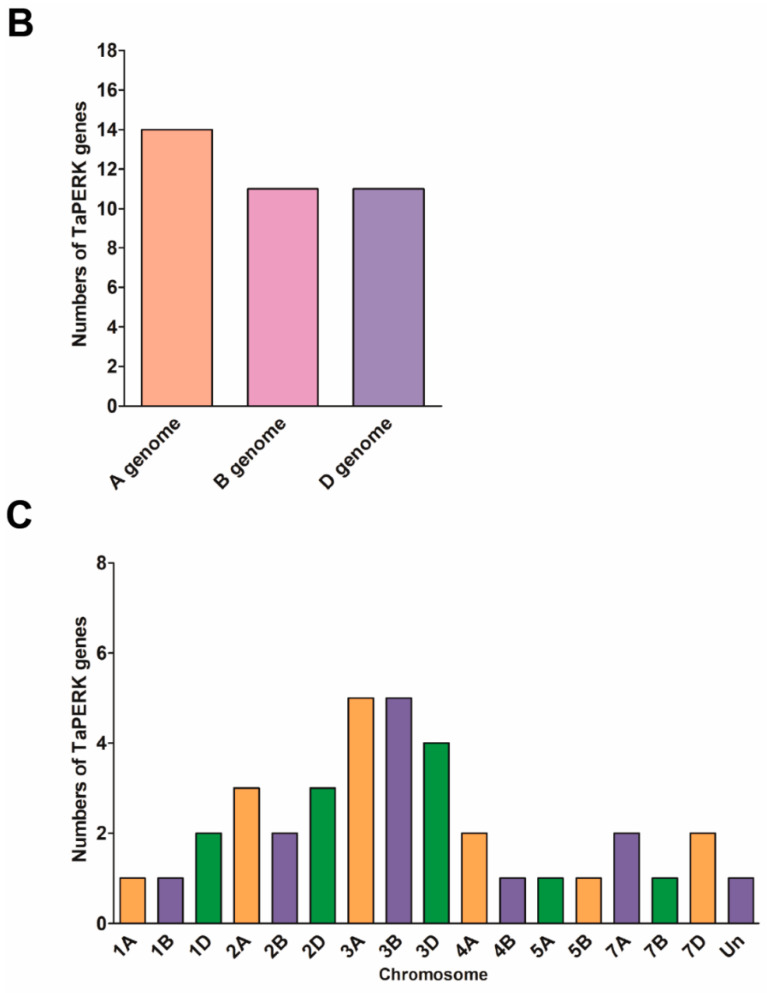
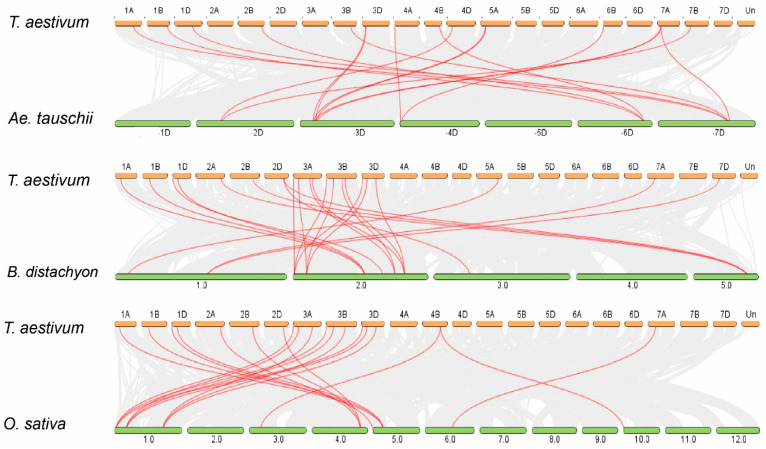
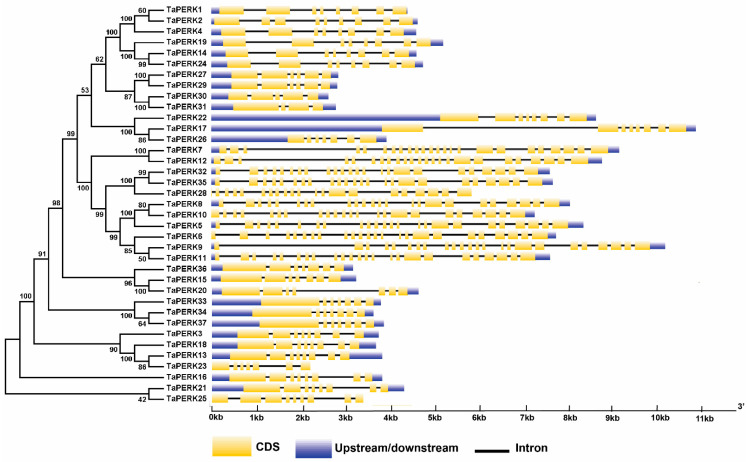
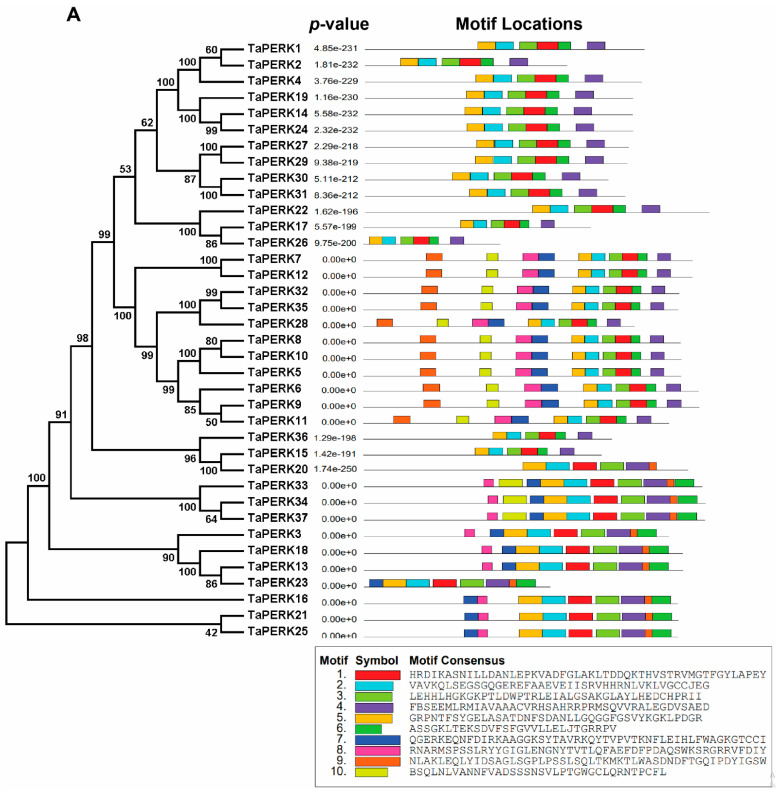
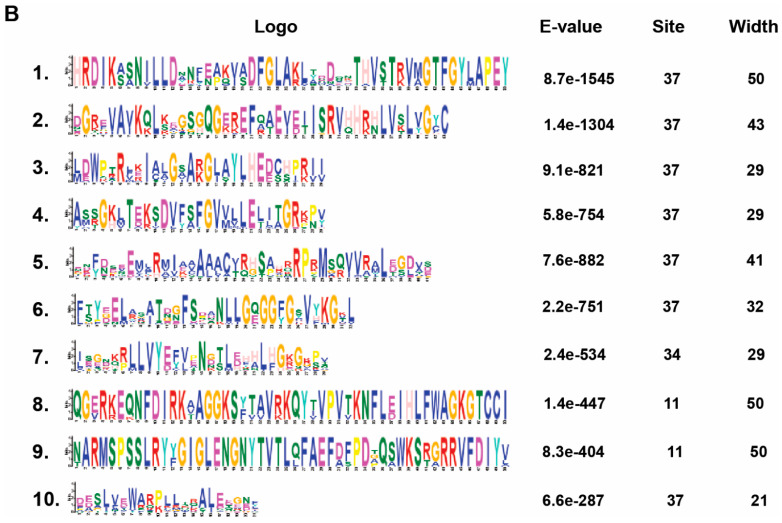
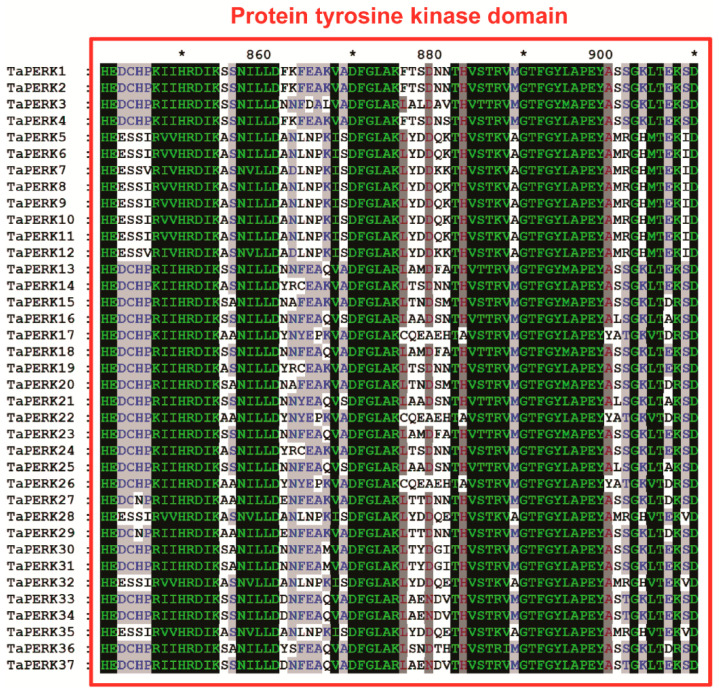
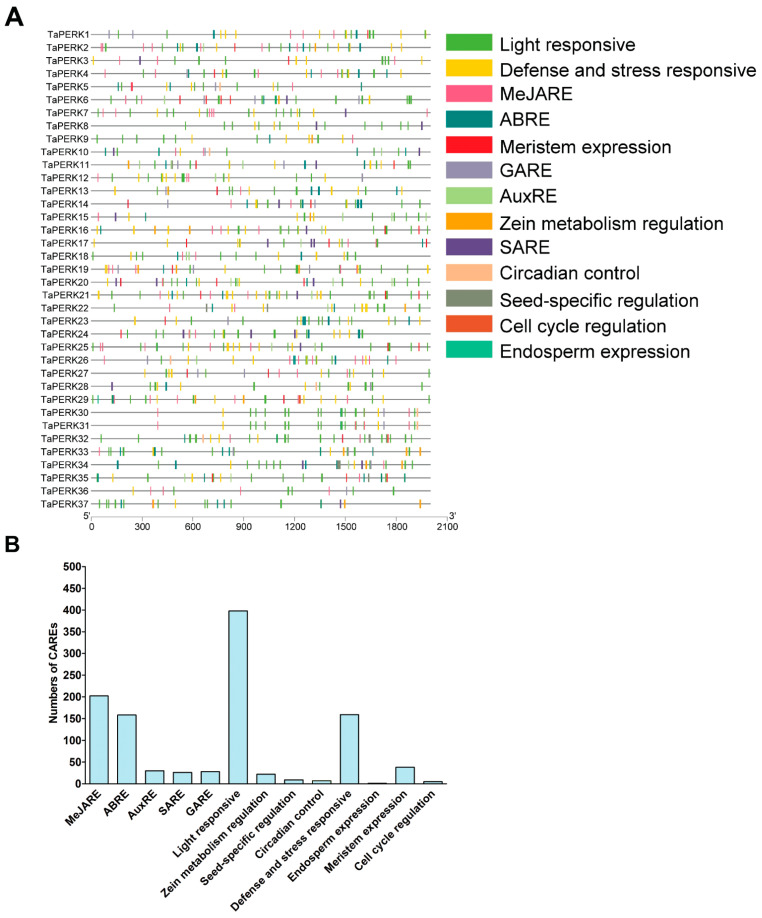
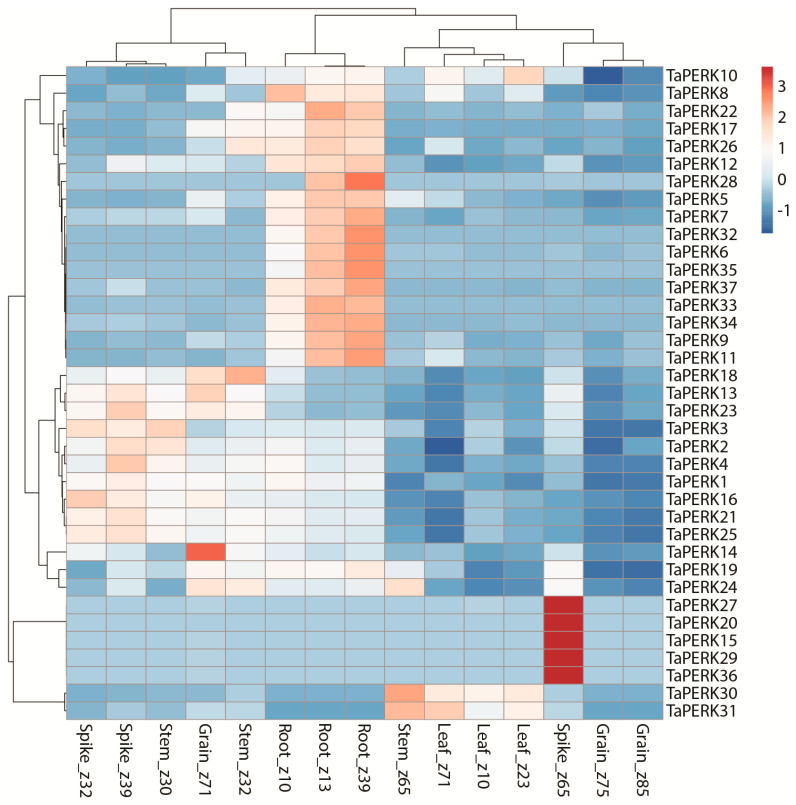
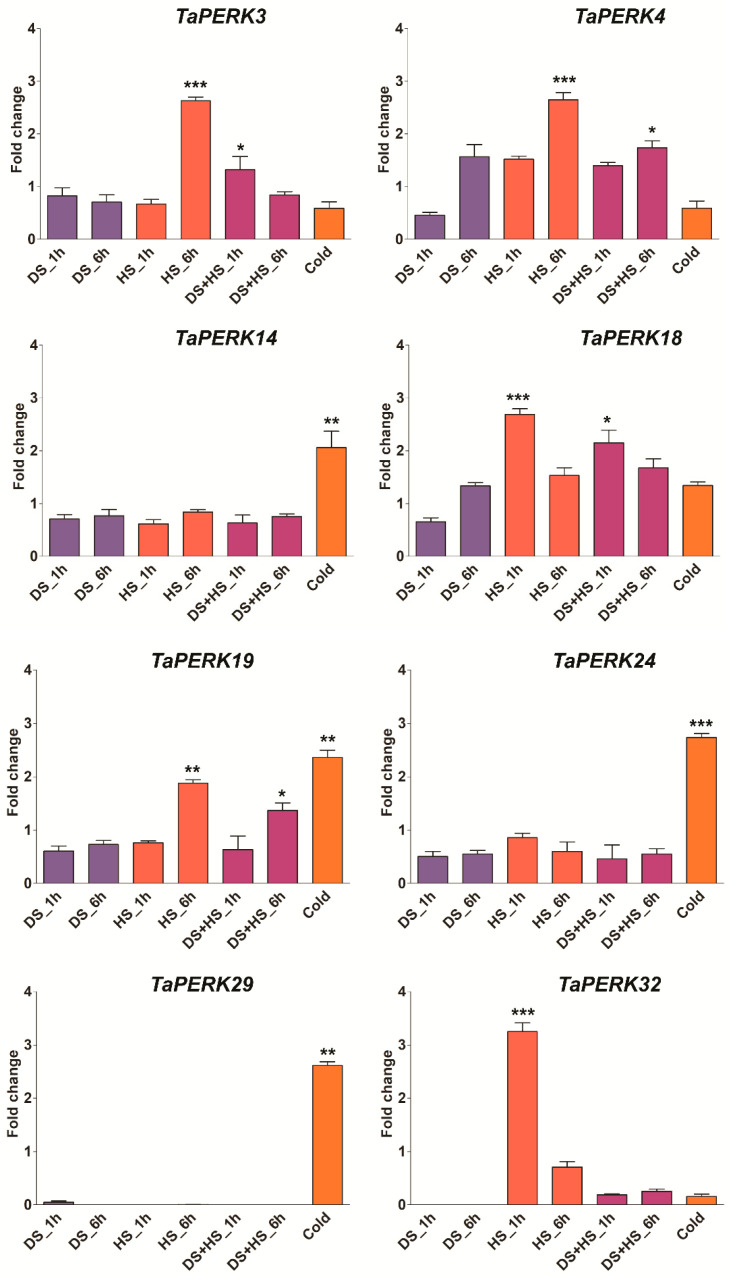
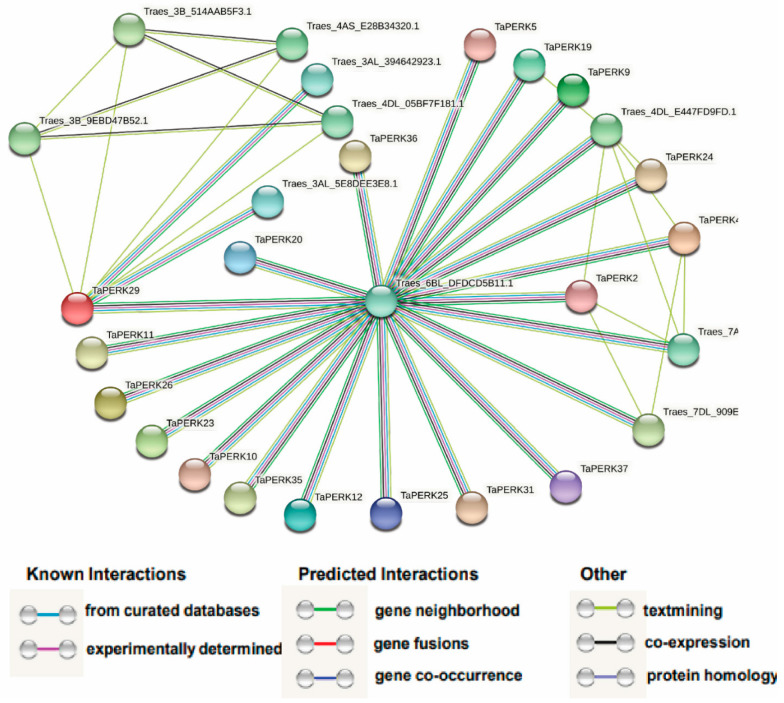
Proline-rich extensin-like receptor kinases (PERKs) are a class of receptor kinases implicated in multiple cellular processes in plants. However, there is a lack of information on the PERK gene family in wheat. Therefore, we identified 37 PERK genes in wheat to understand their role in various developmental processes and stress conditions. Phylogenetic analysis of PERK genes from Arabidopsis thaliana, Oryza sativa, Glycine max, and T. aestivum grouped them into eight well-defined classes. Furthermore, synteny analysis revealed 275 orthologous gene pairs in B. distachyon, Ae. tauschii, T. dicoccoides, O. sativa and A. thaliana. Ka/Ks values showed that most TaPERK genes, except TaPERK1, TaPERK2, TaPERK17, and TaPERK26, underwent strong purifying selection during evolutionary processes. Several cis-acting regulatory elements, essential for plant growth and development and the response to light, phytohormones, and diverse biotic and abiotic stresses, were predicted in the promoter regions of TaPERK genes. In addition, the expression profile of the TaPERK gene family revealed differential expression of TaPERK genes in various tissues and developmental stages. Furthermore, TaPERK gene expression was induced by various biotic and abiotic stresses. The RT-qPCR analysis also revealed similar results with slight variation. Therefore, this study's outcome provides valuable information for elucidating the precise functions of TaPERK in developmental processes and diverse stress conditions in wheat.
Keywords: PERK; RT-qPCR; drought; heat stress; kinase; promoter.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


References
-
- Sim J.-S., Kesawat M.S., Kumar M., Kim S.-Y., Mani V., Subramanian P., Park S., Lee C.-M., Kim S.-R., Hahn B.-S. Lack of the α1, 3-fucosyltransferase gene (OsFucT) affects anther development and pollen viability in rice. Int. J. Mol. Sci. 2018;19:1225. doi: 10.3390/ijms19041225. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
