

Phenotypic Heterogeneity of Variably Protease-Sensitive Prionopathy: A Report of Three Cases Carrying Different Genotypes at PRNP Codon 129
- PMID: 35215959
- PMCID: PMC8879235
- DOI: 10.3390/v14020367
Phenotypic Heterogeneity of Variably Protease-Sensitive Prionopathy: A Report of Three Cases Carrying Different Genotypes at PRNP Codon 129
Abstract
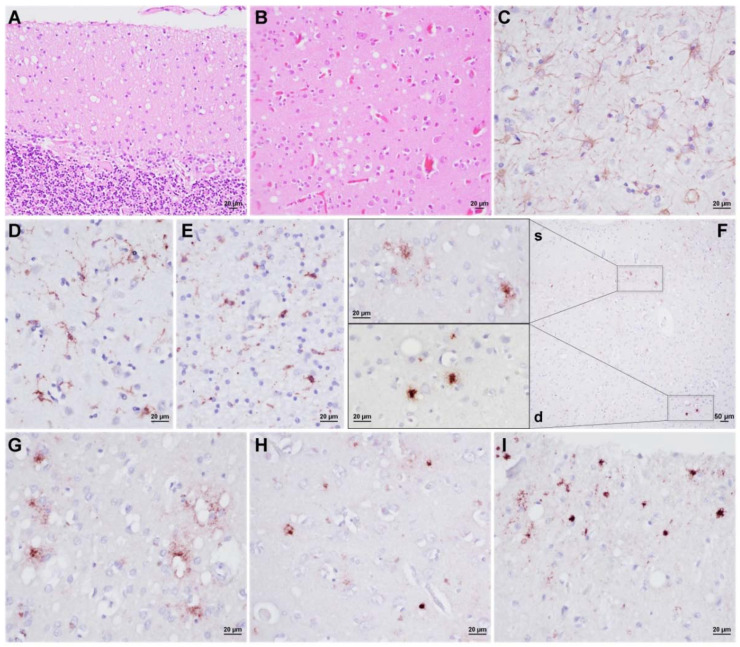
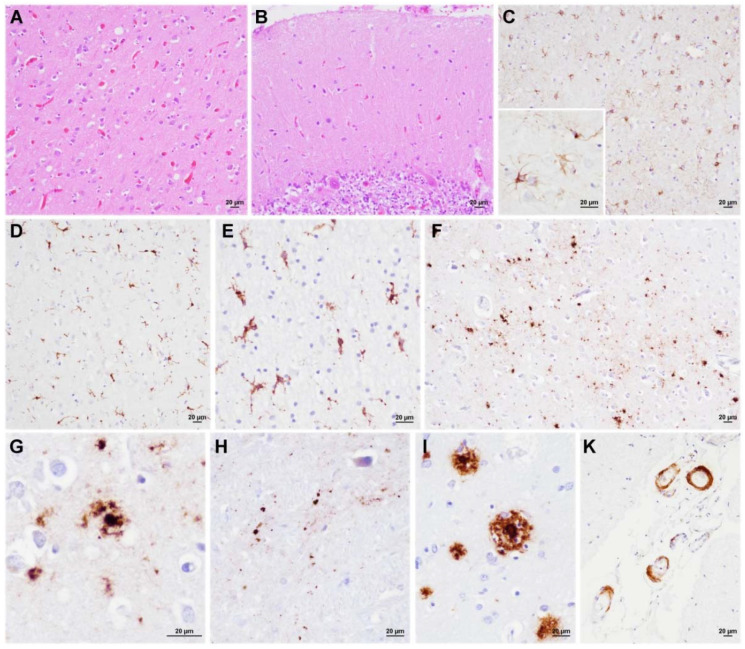
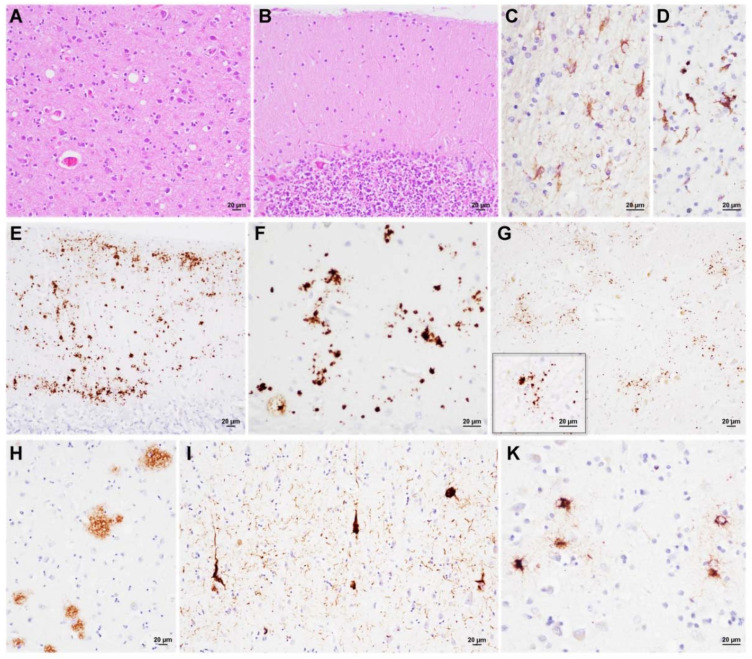
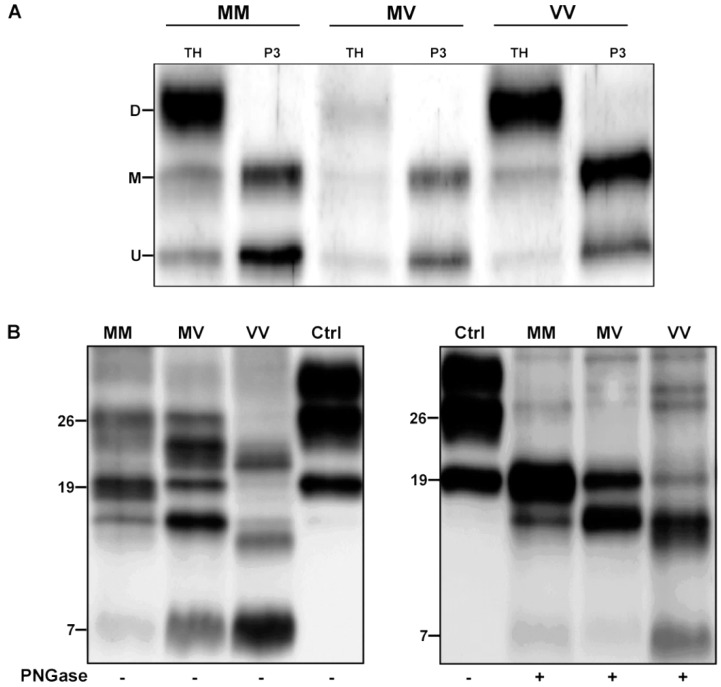
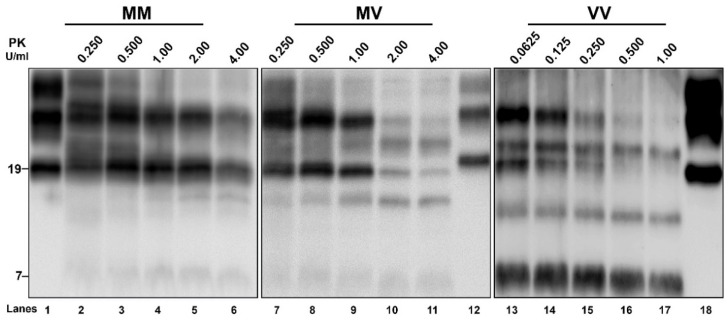
Variably protease-sensitive prionopathy is an exceedingly rare, likely underestimated, sporadic prion disease that is characterized by heterogeneous and often non-specific clinical and pathological features posing diagnostic challenges. We report the results of a comprehensive analysis of three emblematic cases carrying different genotypes at the methionine (M)/valine (V) polymorphic codon 129 in the prion protein gene (PRNP). Clinical, biochemical, and neuropathological findings highlighted the prominent role of the host genetic background as a phenotypic modulator. In particular, the PRNP codon 129 showed a remarkable influence on the physicochemical properties of the pathological prion protein (PrPSc), especially on the sensitivity to proteinase K (PK) digestion (VV > MV > MM), which variably affected the three main fragments (i.e., of 19, 17, and 7 kDa, respectively) comprising the PrPSc profile after PK digestion and immunoblotting. This, in turn, correlated with significant differences in the ratio between the 19 kDa and the 7 kDa fragments which was highest in the MM case and lowest in the VV one. The relative amount of cerebral and cerebellar PrP mini-plaques immunohistochemistry showed a similar association with the codon 129 genotype (i.e., VV > MV > MM). Clinical manifestations and results of diagnostic investigations were non-specific, except for the detection of prion seeding activity by the real-time quaking-induced conversion assay in the only cerebrospinal fluid sample that we tested (from patient 129VV).
Keywords: CJD; Creutzfeldt-Jakob disease; PRNP; RT-QuIC; VPSPr; amyloid; prion disease; protein misfolding; real-time quaking-induced conversion; scrapie.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Characterization of variably protease-sensitive prionopathy by capillary electrophoresis.Sci Rep. 2024 Nov 13;14(1):27867. doi: 10.1038/s41598-024-79217-1. Sci Rep. 2024. PMID: 39537719 Free PMC article.
-
A novel form of human disease with a protease-sensitive prion protein and heterozygosity methionine/valine at codon 129: Case report.BMC Neurol. 2010 Oct 25;10:99. doi: 10.1186/1471-2377-10-99. BMC Neurol. 2010. PMID: 20973975 Free PMC article.
-
The prion protein protease sensitivity, stability and seeding activity in variably protease sensitive prionopathy brain tissue suggests molecular overlaps with sporadic Creutzfeldt-Jakob disease.Acta Neuropathol Commun. 2014 Oct 21;2:152. doi: 10.1186/s40478-014-0152-4. Acta Neuropathol Commun. 2014. PMID: 25331173 Free PMC article.
-
Intracerebral distribution of the abnormal isoform of the prion protein in sporadic Creutzfeldt-Jakob disease and fatal insomnia.Microsc Res Tech. 2000 Jul 1;50(1):16-25. doi: 10.1002/1097-0029(20000701)50:1<16::AID-JEMT4>3.0.CO;2-Y. Microsc Res Tech. 2000. PMID: 10871544 Review.
-
Neuropathological and biochemical criteria to identify acquired Creutzfeldt-Jakob disease among presumed sporadic cases.Neuropathology. 2016 Jun;36(3):305-10. doi: 10.1111/neup.12270. Epub 2015 Dec 15. Neuropathology. 2016. PMID: 26669818 Review.
Cited by
-
Human prion diseases and the prion protein - what is the current state of knowledge?Transl Neurosci. 2023 Oct 16;14(1):20220315. doi: 10.1515/tnsci-2022-0315. eCollection 2023 Jan 1. Transl Neurosci. 2023. PMID: 37854584 Free PMC article. Review.
-
Variably protease-sensitive prionopathy with methionine homozygosity at codon 129 in the prion protein gene.BMJ Case Rep. 2024 Feb 22;17(2):e258199. doi: 10.1136/bcr-2023-258199. BMJ Case Rep. 2024. PMID: 38388201
-
The Use of Real-Time Quaking-Induced Conversion for the Diagnosis of Human Prion Diseases.Front Aging Neurosci. 2022 Apr 25;14:874734. doi: 10.3389/fnagi.2022.874734. eCollection 2022. Front Aging Neurosci. 2022. PMID: 35547619 Free PMC article. Review.
-
Characterization of variably protease-sensitive prionopathy by capillary electrophoresis.Sci Rep. 2024 Nov 13;14(1):27867. doi: 10.1038/s41598-024-79217-1. Sci Rep. 2024. PMID: 39537719 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
