

Interaction between the Hepatitis B Virus and Cellular FLIP Variants in Viral Replication and the Innate Immune System
- PMID: 35215970
- PMCID: PMC8874586
- DOI: 10.3390/v14020373
Interaction between the Hepatitis B Virus and Cellular FLIP Variants in Viral Replication and the Innate Immune System
Abstract
During viral evolution and adaptation, many viruses have utilized host cellular factors and machinery as their partners. HBx, as a multifunctional viral protein encoded by the hepatitis B virus (HBV), promotes HBV replication and greatly contributes to the development of HBV-associated hepatocellular carcinoma (HCC). HBx interacts with several host factors in order to regulate HBV replication and evolve carcinogenesis. The cellular FADD-like IL-1β-converting enzyme (FLICE)-like inhibitory protein (c-FLIP) is a major factor that functions in a variety of cellular pathways and specifically in apoptosis. It has been shown that the interaction between HBx and c-FLIP determines HBV fate. In this review, we provide a comprehensive and detailed overview of the interplay between c-FLIP and HBV in various environmental circumstances. We describe strategies adapted by HBV to establish its chronic infection. We also summarize the conventional roles of c-FLIP and highlight the functional outcome of the interaction between c-FLIP and HBV or other viruses in viral replication and the innate immune system.
Keywords: HBx; cellular FLIP (c-FLIP); hepatitis B virus; innate immune system; viral FLIP (v-FLIP).
Conflict of interest statement
The authors declare no conflict of interest.
Figures
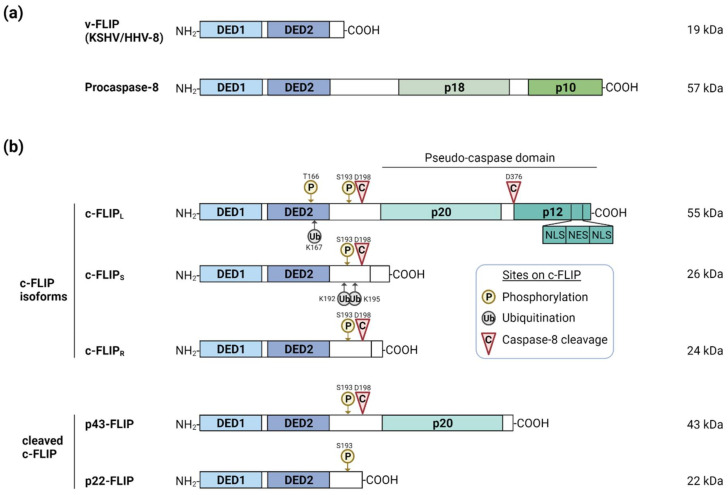
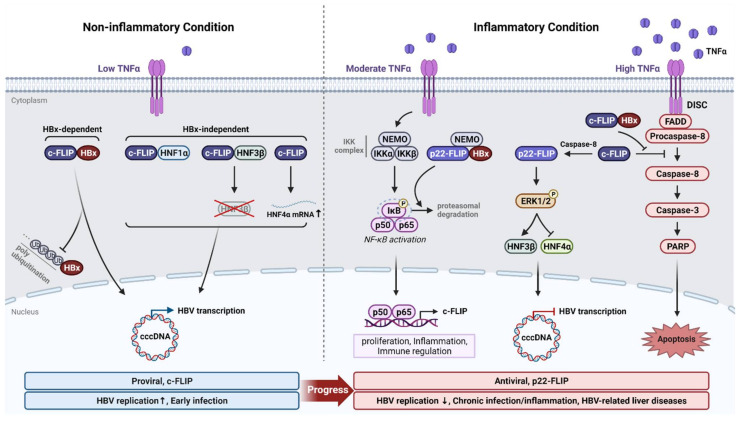
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
