

A blood atlas of COVID-19 defines hallmarks of disease severity and specificity
- PMID: 35216673
- PMCID: PMC8776501
- DOI: 10.1016/j.cell.2022.01.012
A blood atlas of COVID-19 defines hallmarks of disease severity and specificity
Abstract
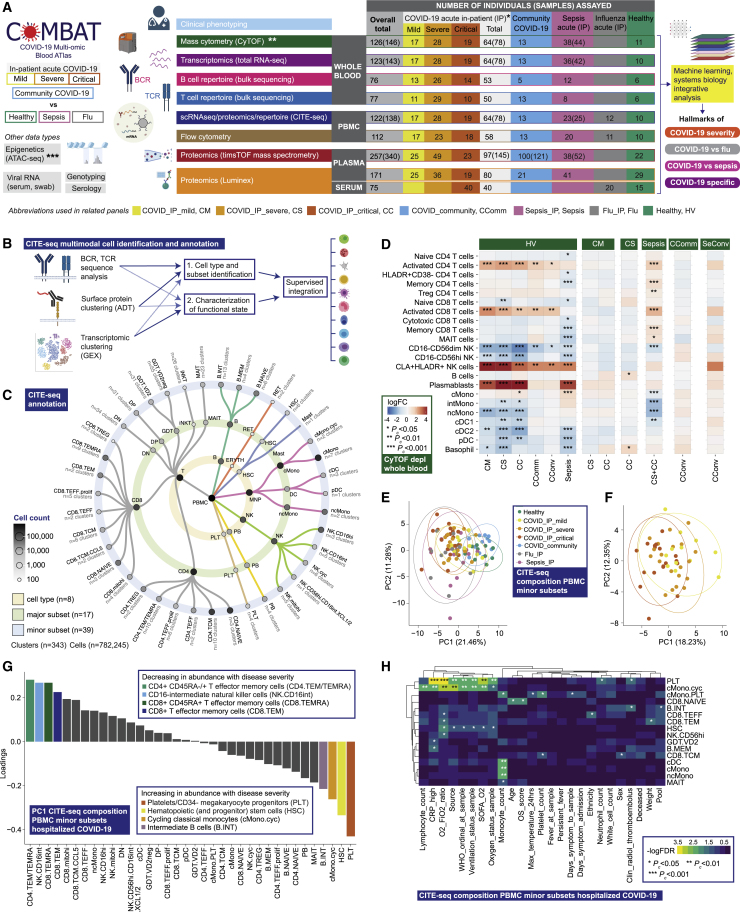
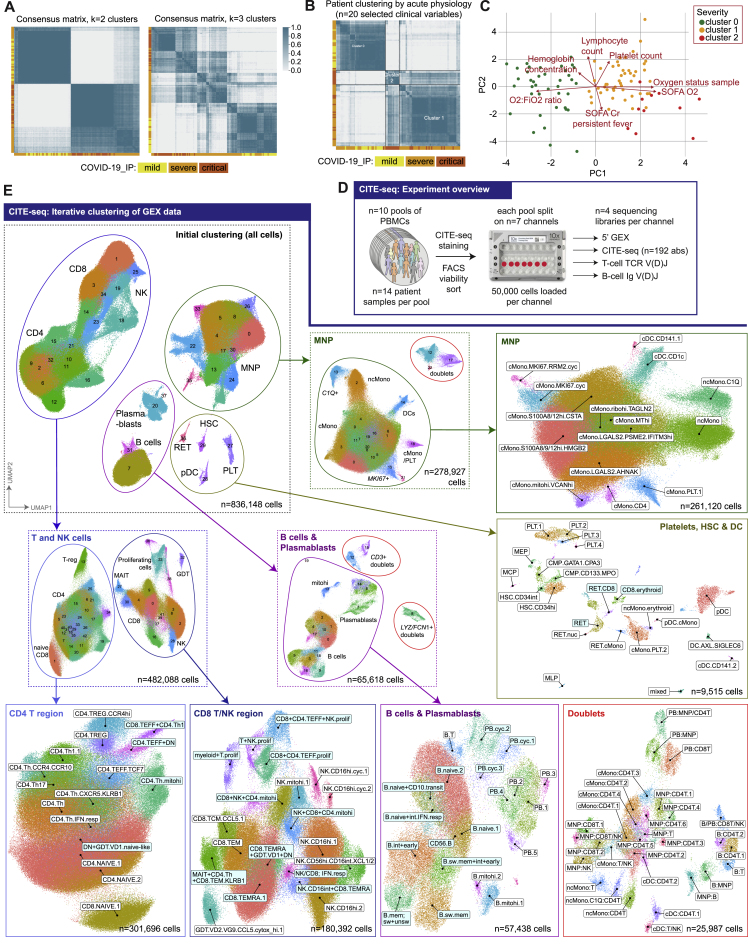
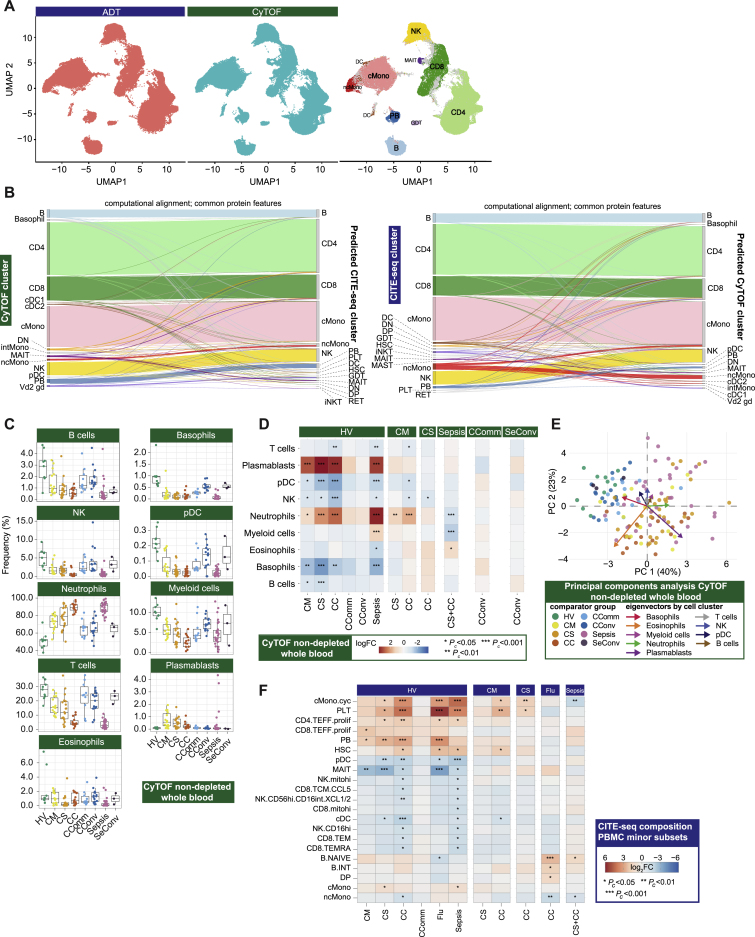
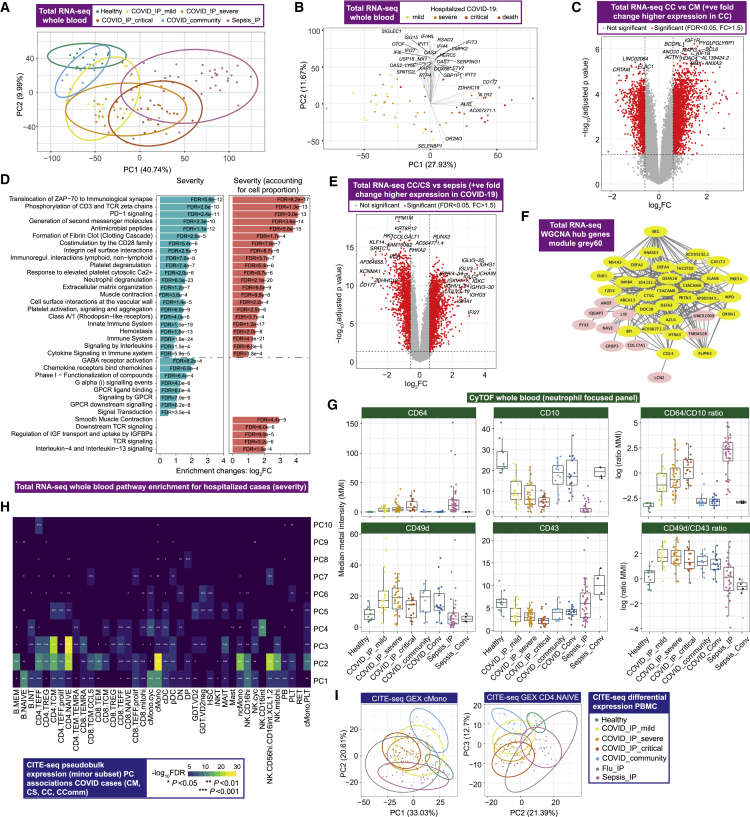
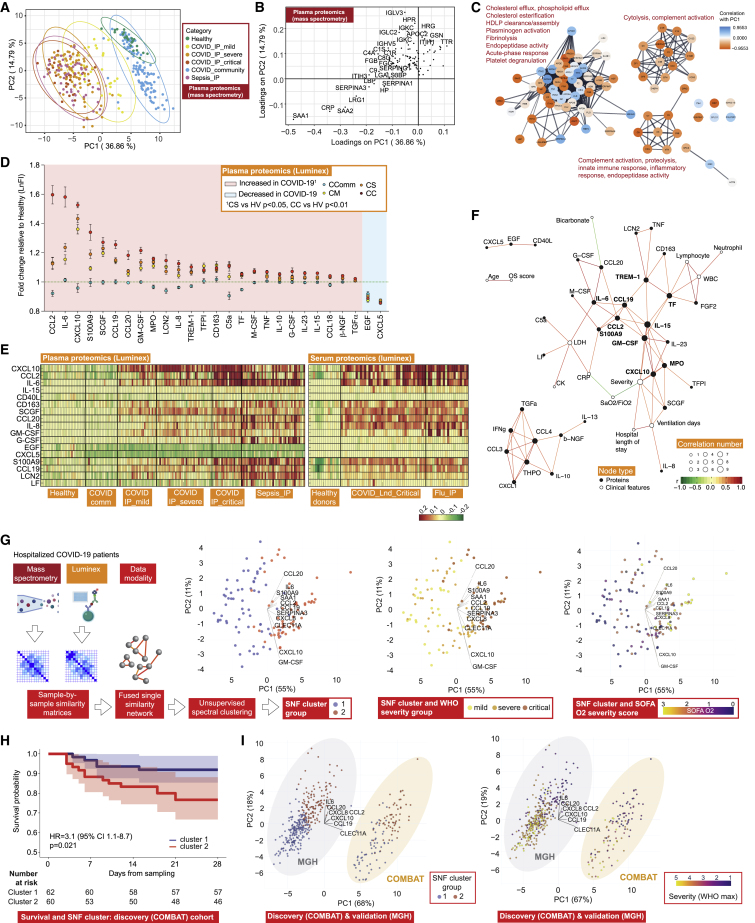
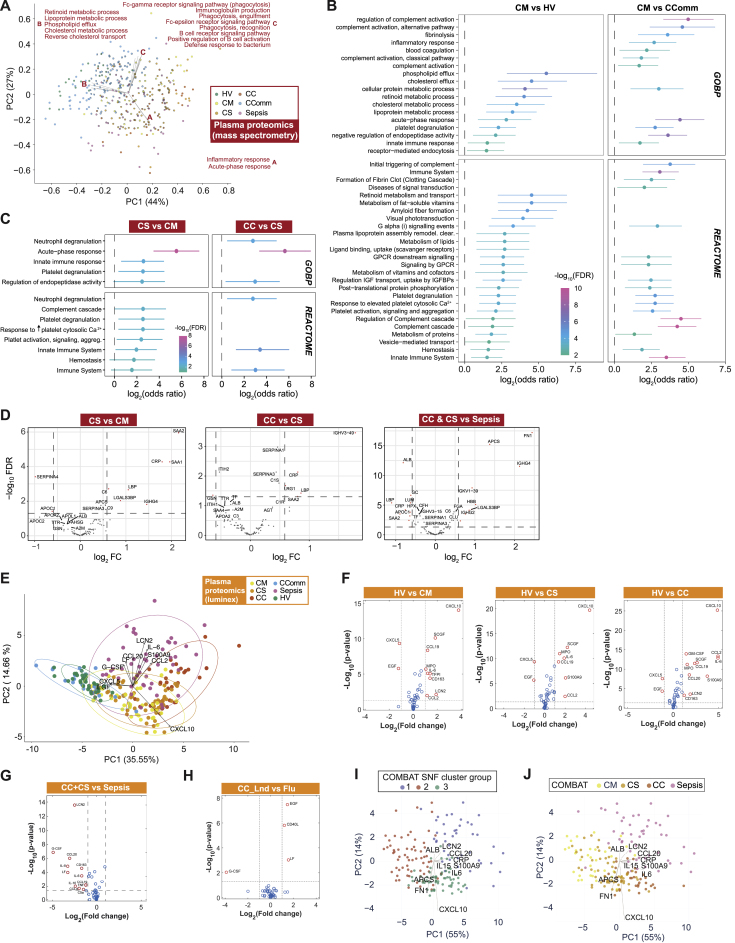
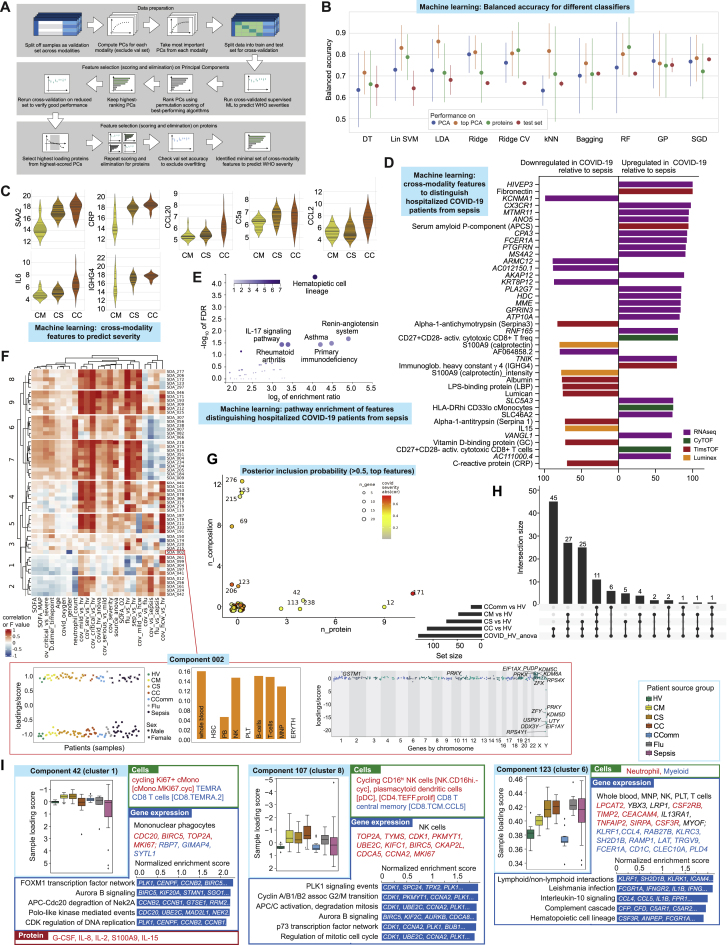
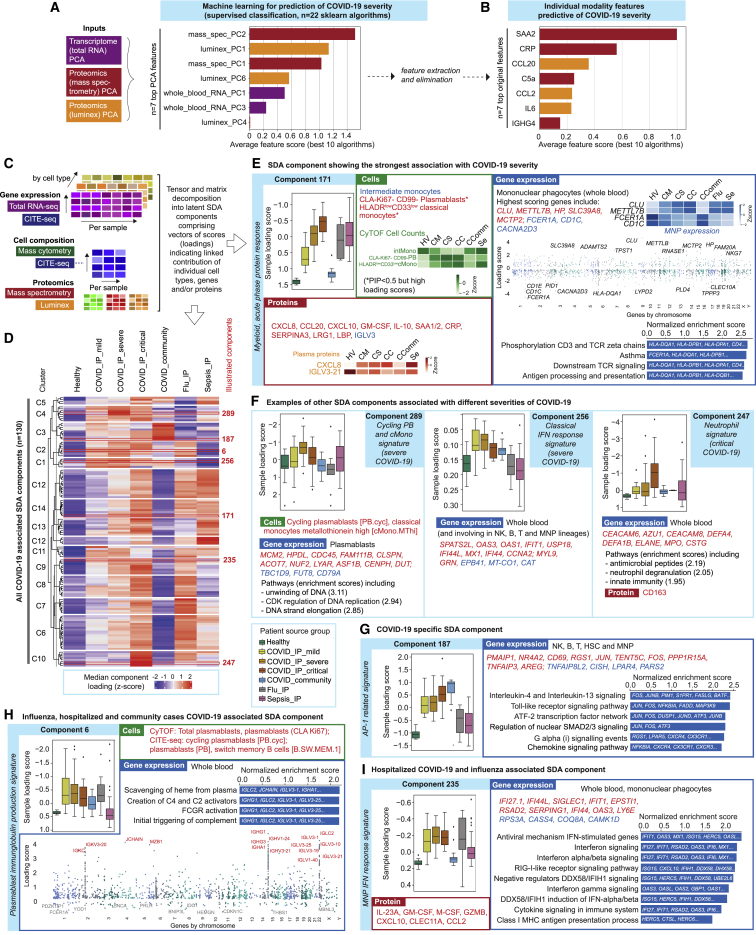
Treatment of severe COVID-19 is currently limited by clinical heterogeneity and incomplete description of specific immune biomarkers. We present here a comprehensive multi-omic blood atlas for patients with varying COVID-19 severity in an integrated comparison with influenza and sepsis patients versus healthy volunteers. We identify immune signatures and correlates of host response. Hallmarks of disease severity involved cells, their inflammatory mediators and networks, including progenitor cells and specific myeloid and lymphocyte subsets, features of the immune repertoire, acute phase response, metabolism, and coagulation. Persisting immune activation involving AP-1/p38MAPK was a specific feature of COVID-19. The plasma proteome enabled sub-phenotyping into patient clusters, predictive of severity and outcome. Systems-based integrative analyses including tensor and matrix decomposition of all modalities revealed feature groupings linked with severity and specificity compared to influenza and sepsis. Our approach and blood atlas will support future drug development, clinical trial design, and personalized medicine approaches for COVID-19.
Keywords: COVID-19; SARS-CoV-2; blood; coronavirus; epigenetics; immune; multi-omics; personalized medicine; proteomics; transcriptomics.
Copyright © 2022 The Author. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests R.B.-R. (co-founder and consultant Alchemab Therapeutics Ltd), R.C. (founder MIROBio), J. Hughes (director and shareholder Nucleome Therapeutics), G.S. (GSK Vaccines SAB), J.A.T. (GSK Human Genetics SAB). Other authors declare no competing interests.
Figures





References
-
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. - PubMed
-
- Andrews S. 2010. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Publication types
MeSH terms
Substances
Grants and funding
- 204290/Z/16/Z/WT_/Wellcome Trust/United Kingdom
- 204969/Z/16/Z/WT_/Wellcome Trust/United Kingdom
- MC_PC_19025/MRC_/Medical Research Council/United Kingdom
- MC_UU_00016/14/MRC_/Medical Research Council/United Kingdom
- MR/S005471/1/MRC_/Medical Research Council/United Kingdom
- MR/X001210/1/MRC_/Medical Research Council/United Kingdom
- 29034/CRUK_/Cancer Research UK/United Kingdom
- 27125/CRUK_/Cancer Research UK/United Kingdom
- MR/S025308/1/MRC_/Medical Research Council/United Kingdom
- MR/S020918/1/MRC_/Medical Research Council/United Kingdom
- G84/6443/MRC_/Medical Research Council/United Kingdom
- MR/T014067/1/MRC_/Medical Research Council/United Kingdom
- MC_UU_00008/10/MRC_/Medical Research Council/United Kingdom
- 28051/CRUK_/Cancer Research UK/United Kingdom
- MC_PC_20002/MRC_/Medical Research Council/United Kingdom
- 26988/CRUK_/Cancer Research UK/United Kingdom
- MC_UU_00008/5/MRC_/Medical Research Council/United Kingdom
- 208750/Z/17/Z/WT_/Wellcome Trust/United Kingdom
- 27723/CRUK_/Cancer Research UK/United Kingdom
- 201488/Z/16/Z/WT_/Wellcome Trust/United Kingdom
- MC_UU_00008/6/MRC_/Medical Research Council/United Kingdom
- 22252/VAC_/Versus Arthritis/United Kingdom
- MR/V010182/1/MRC_/Medical Research Council/United Kingdom
- MR/L006340/1/MRC_/Medical Research Council/United Kingdom
- MC_PC_19059/MRC_/Medical Research Council/United Kingdom
- R24 DK106766/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
