

Conservation of chromatin conformation in carnivores
- PMID: 35217621
- PMCID: PMC8892538
- DOI: 10.1073/pnas.2120555119
Conservation of chromatin conformation in carnivores
Abstract
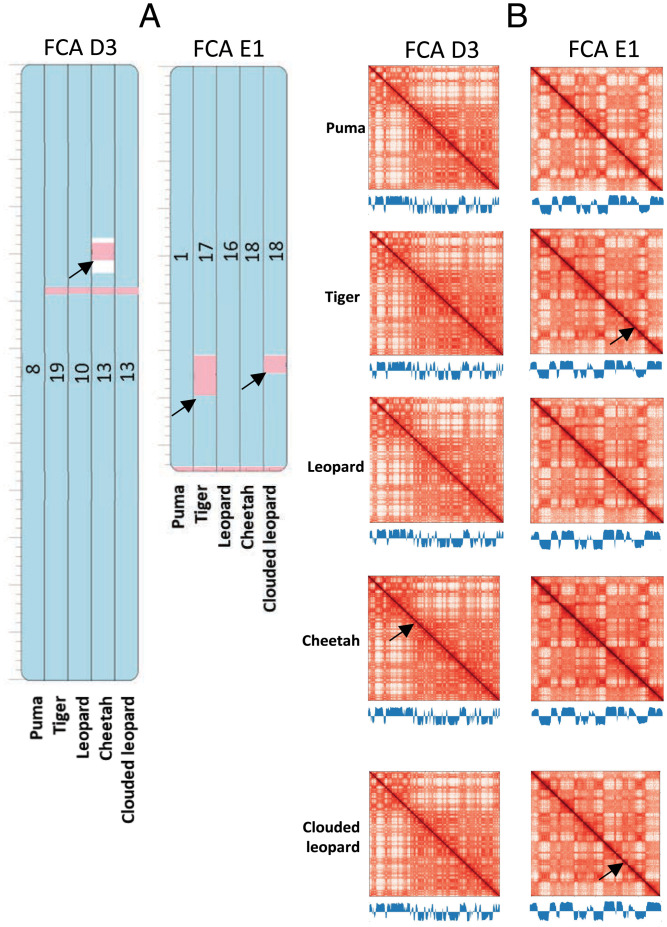
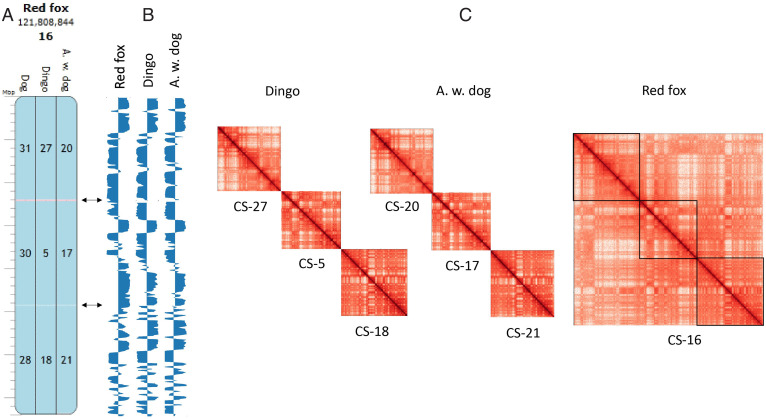
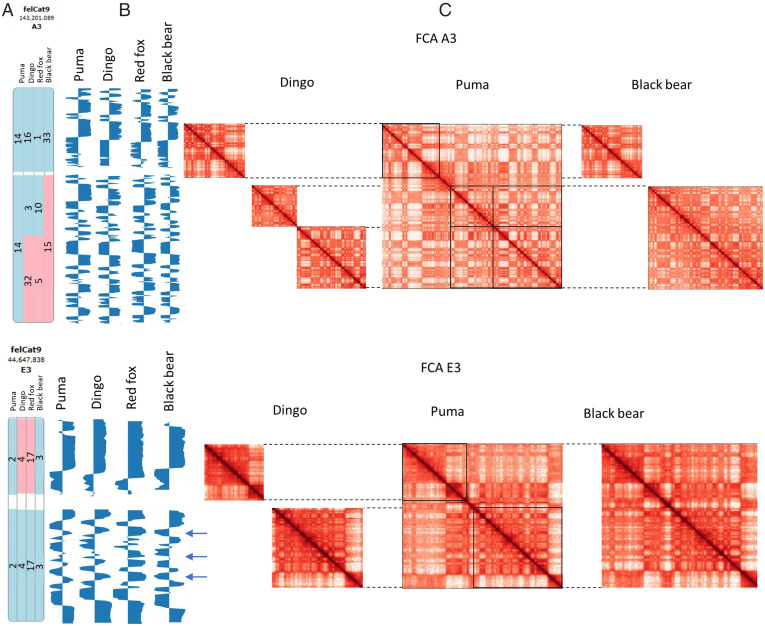
High throughput chromatin conformation capture (Hi-C) of leukocyte DNA was used to investigate the evolutionary stability of chromatin conformation at the chromosomal level in 11 species from three carnivore families: Felidae, Canidae, and Ursidae. Chromosome-scale scaffolds (C-scaffolds) of each species were initially used for whole-genome alignment to a reference genome within each family. This approach established putative orthologous relationships between C-scaffolds among the different species. Hi-C contact maps for all C-scaffolds were then visually compared and found to be distinct for a given reference chromosome or C-scaffold within a species and indistinguishable for orthologous C-scaffolds having a 1:1 relationship within a family. The visual patterns within families were strongly supported by eigenvectors from the Hi-C contact maps. Analysis of Hi-C contact maps and eigenvectors across the three carnivore families revealed that most cross-family orthologous subchromosomal fragments have a conserved three-dimensional (3D) chromatin structure and thus have been under strong evolutionary constraint for ∼54 My of carnivore evolution. The most pronounced differences in chromatin conformation were observed for the X chromosome and the red fox genome, whose chromosomes have undergone extensive rearrangements relative to other canids. We also demonstrate that Hi-C contact map pattern analysis can be used to accurately identify orthologous relationships between C-scaffolds and chromosomes, a method we termed "3D comparative scaffotyping." This method provides a powerful means for estimating karyotypes in de novo sequenced species that have unknown karyotype and no physical mapping information.
Keywords: carnivores; chromatin conformation; chromosome evolution; mammals.
Copyright © 2022 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no competing interest.
Figures
References
-
- Cremer T., Cremer C., Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet. 2, 292–301 (2001). - PubMed
