

New treatment strategies for advanced-stage gastrointestinal stromal tumours
- PMID: 35217782
- PMCID: PMC11488293
- DOI: 10.1038/s41571-022-00606-4
New treatment strategies for advanced-stage gastrointestinal stromal tumours
Abstract
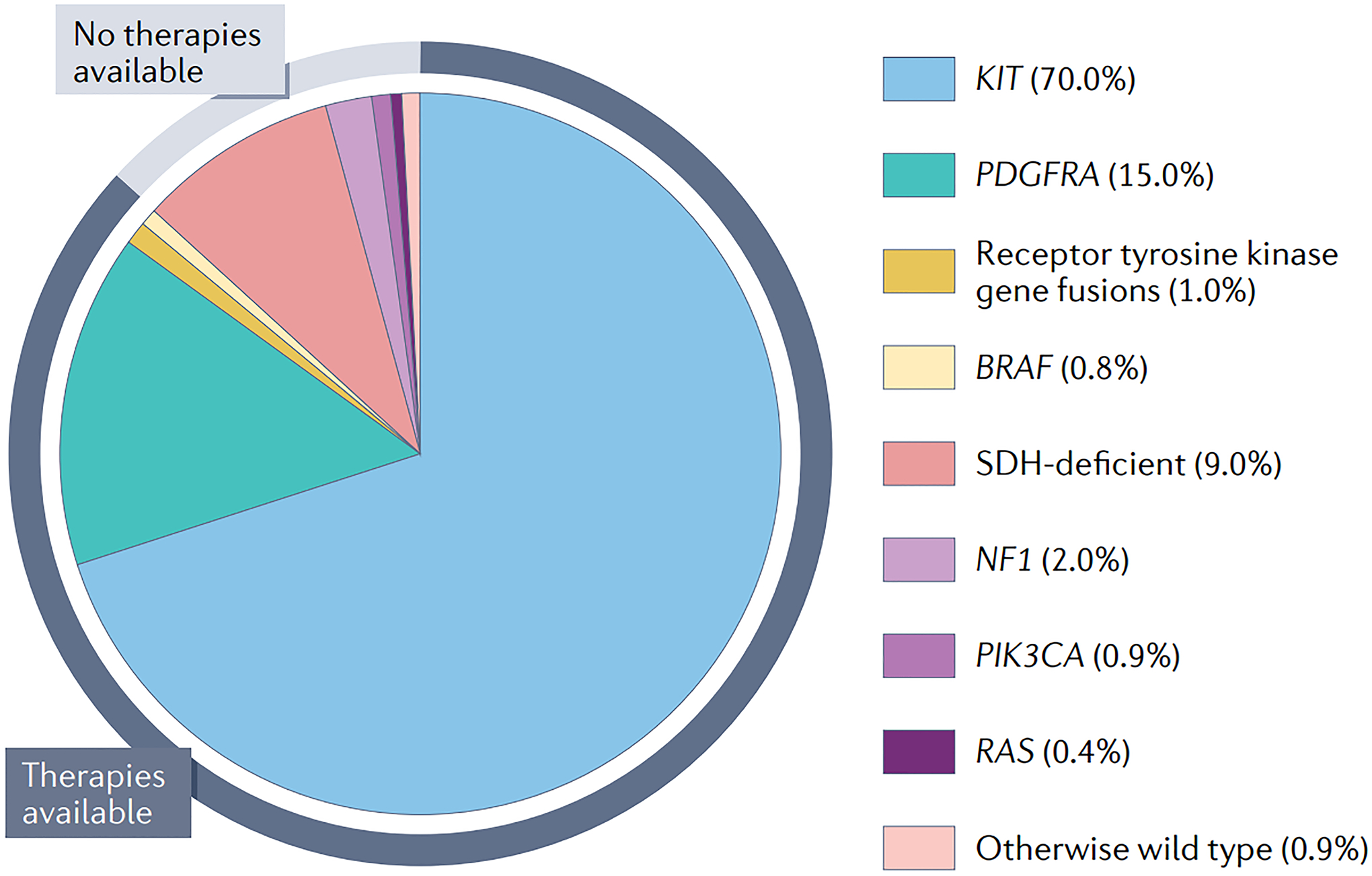
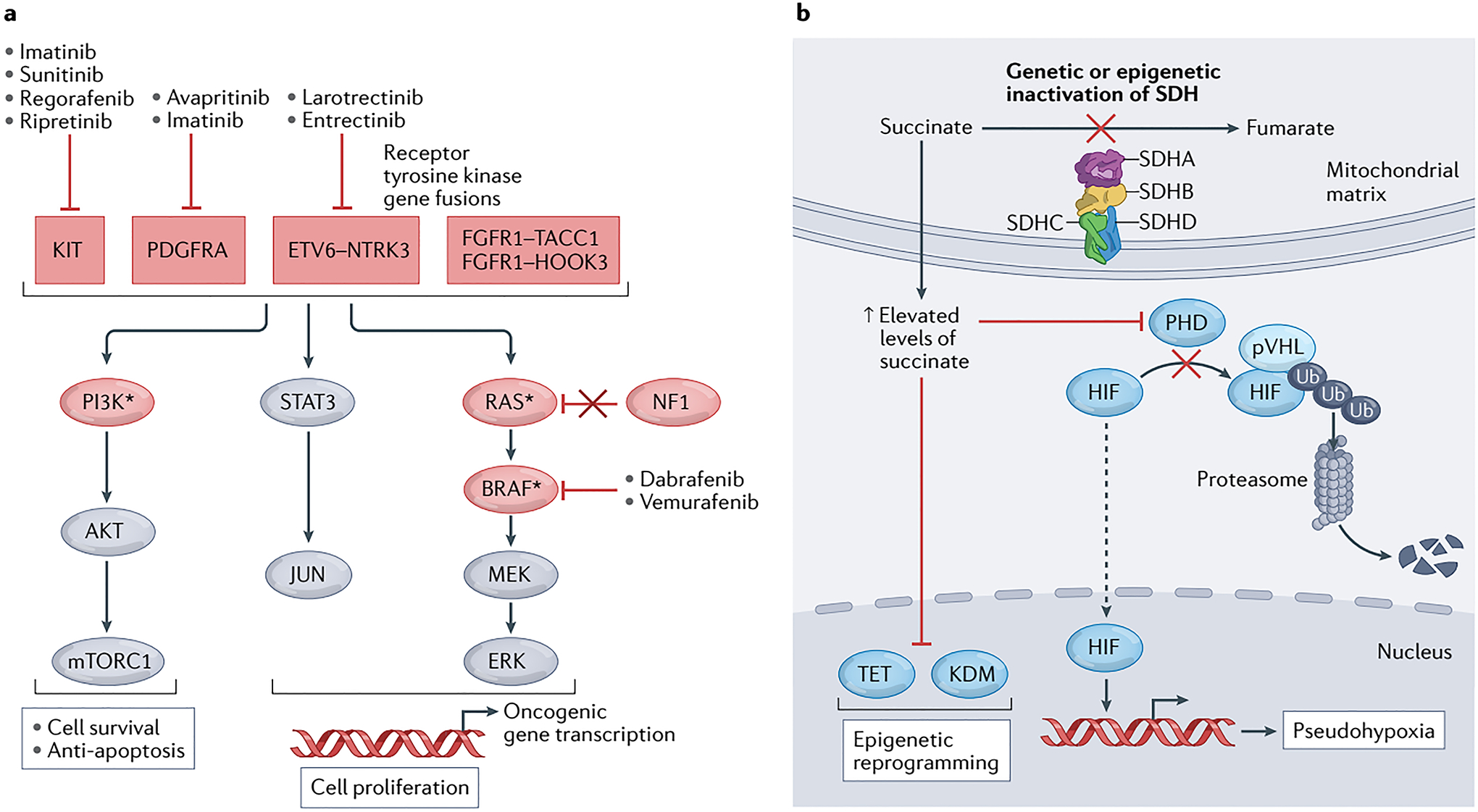
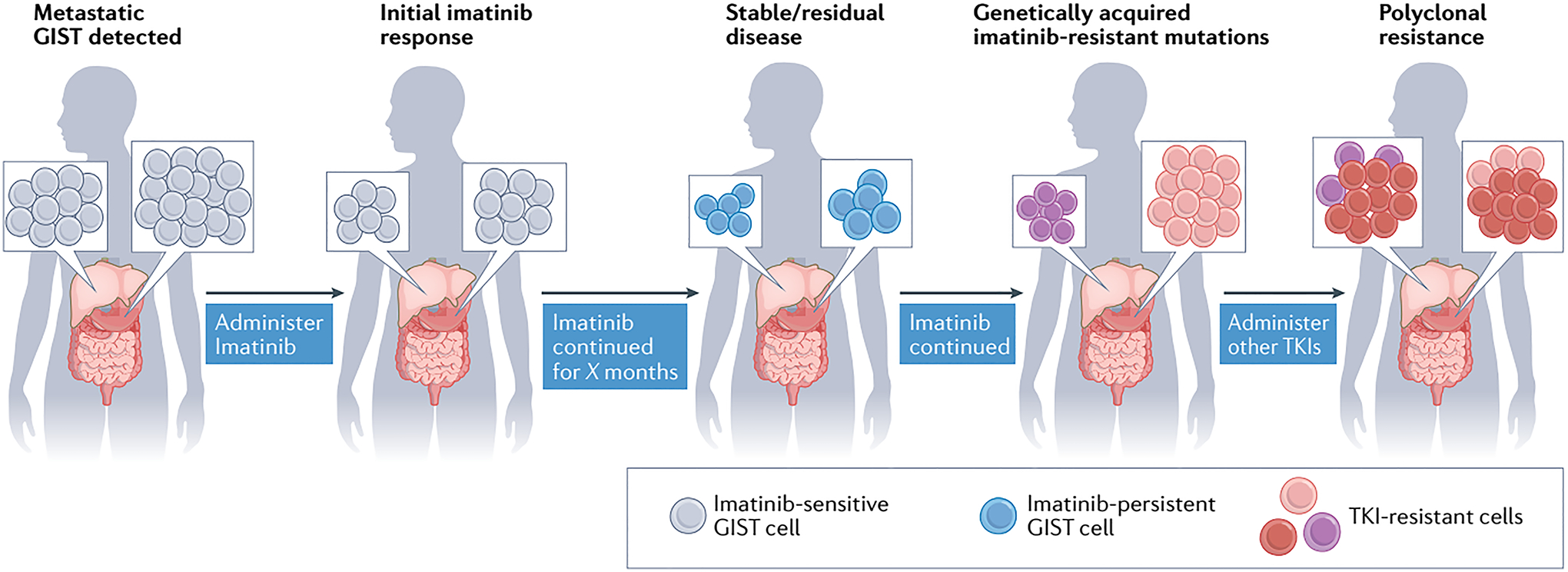
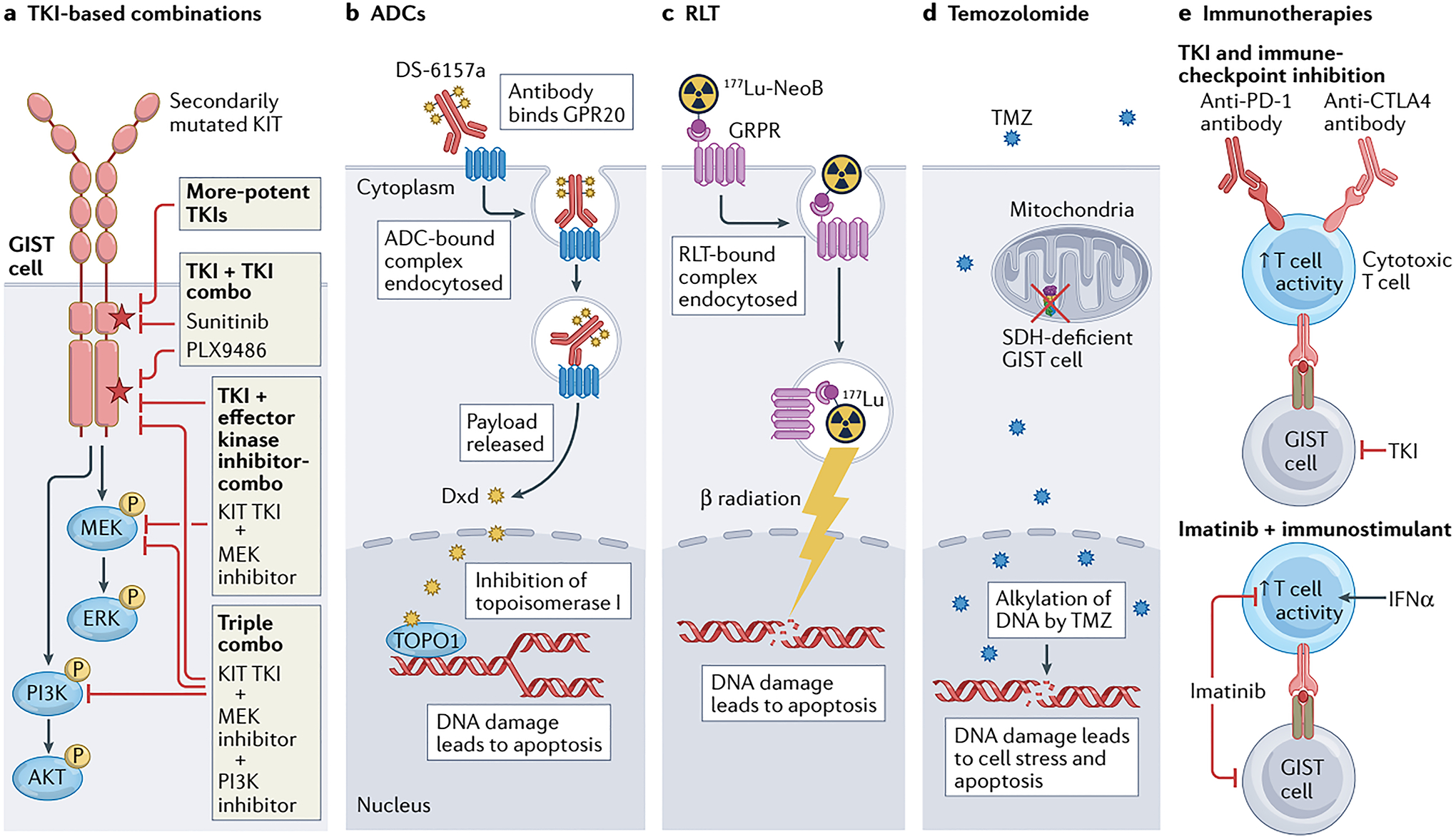
When gastrointestinal stromal tumour (GIST), the most common form of sarcoma, was first recognized as a distinct pathological entity in the 1990s, patients with advanced-stage disease had a very poor prognosis owing to a lack of effective medical therapies. The discovery of KIT mutations as the first and most prevalent drivers of GIST and the subsequent development of the first KIT tyrosine kinase inhibitor (TKI), imatinib, revolutionized the treatment of patients with this disease. We can now identify the driver mutation in 99% of patients with GIST via molecular diagnostic testing, and therapies have been developed to treat many, but not all, molecular subtypes of the disease. At present, seven drugs are approved by the FDA for the treatment of advanced-stage GIST (imatinib, sunitinib, regorafenib, ripretinib, avapritinib, larotrectinib and entrectinib), all of which are TKIs. Although these agents can be very effective for treating certain GIST subtypes, challenges remain and new therapeutic approaches are needed. In this Review, we discuss the molecular subtypes of GIST and the evolution of current treatments, as well as their therapeutic limitations. We also highlight emerging therapeutic approaches that might overcome clinical challenges through novel strategies predicated on the biological features of the distinct GIST molecular subtypes.
© 2022. This is a U.S. government work and not under copyright protection in the U.S.; foreign copyright protection may apply.
Conflict of interest statement
Competing interests
M.C.H. has been a consultant for Blueprint Medicines, Deciphera Pharmaceuticals, Novartis and Theseus Pharmaceuticals, and has a patent for the treatment of GIST using imatinib that has been licensed by his institute to Novartis. The other authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
