

Bile acid and receptors: biology and drug discovery for nonalcoholic fatty liver disease
- PMID: 35217817
- PMCID: PMC9061718
- DOI: 10.1038/s41401-022-00880-z
Bile acid and receptors: biology and drug discovery for nonalcoholic fatty liver disease
Abstract
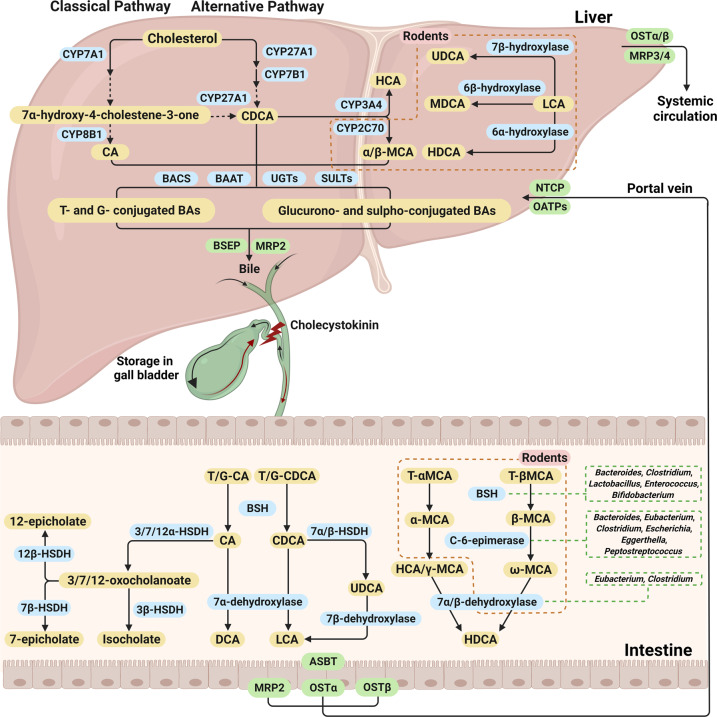
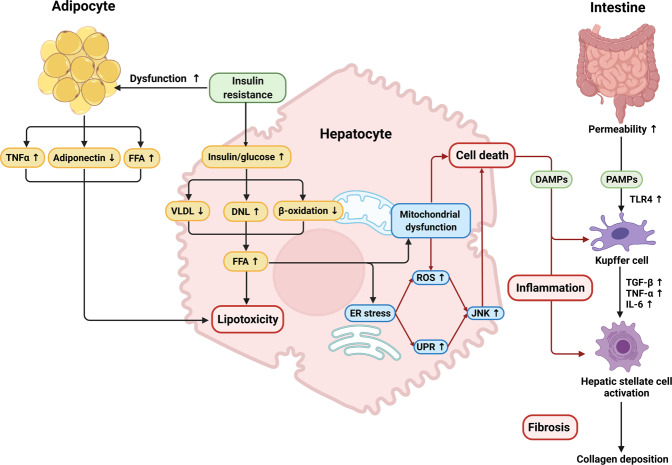
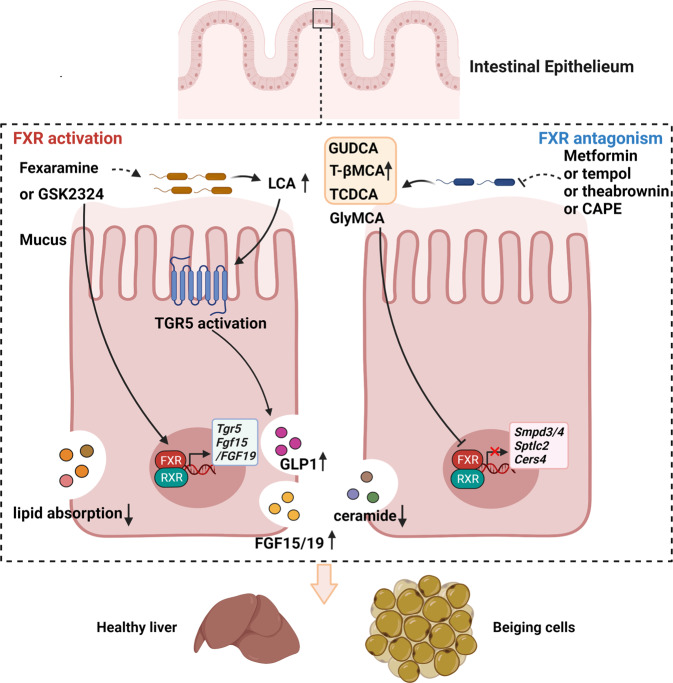
Nonalcoholic fatty liver disease (NAFLD), a series of liver metabolic disorders manifested by lipid accumulation within hepatocytes, has become the primary cause of chronic liver diseases worldwide. About 20%-30% of NAFLD patients advance to nonalcoholic steatohepatitis (NASH), along with cell death, inflammation response and fibrogenesis. The pathogenesis of NASH is complex and its development is strongly related to multiple metabolic disorders (e.g. obesity, type 2 diabetes and cardiovascular diseases). The clinical outcomes include liver failure and hepatocellular cancer. There is no FDA-approved NASH drug so far, and thus effective therapeutics are urgently needed. Bile acids are synthesized in hepatocytes, transported into the intestine, metabolized by gut bacteria and recirculated back to the liver by the enterohepatic system. They exert pleiotropic roles in the absorption of fats and regulation of metabolism. Studies on the relevance of bile acid disturbance with NASH render it as an etiological factor in NASH pathogenesis. Recent findings on the functional identification of bile acid receptors have led to a further understanding of the pathophysiology of NASH such as metabolic dysregulation and inflammation, and bile acid receptors are recognized as attractive targets for NASH treatment. In this review, we summarize the current knowledge on the role of bile acids and the receptors in the development of NAFLD and NASH, especially the functions of farnesoid X receptor (FXR) in different tissues including liver and intestine. The progress in the development of bile acid and its receptors-based drugs for the treatment of NASH including bile acid analogs and non-bile acid modulators on bile acid metabolism is also discussed.
Keywords: Farnesoid X receptor; G protein-coupled bile acid receptor; bile acids; drug target; nonalcoholic fatty liver disease; nonalcoholic steatohepatitis.
© 2022. The Author(s), under exclusive licence to Shanghai Institute of Materia Medica, Chinese Academy of Sciences and Chinese Pharmacological Society.
Conflict of interest statement
The authors declare no competing interests.
Figures

Similar articles
-
The role of bile acids in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis.Mol Aspects Med. 2017 Aug;56:34-44. doi: 10.1016/j.mam.2017.04.004. Epub 2017 May 5. Mol Aspects Med. 2017. PMID: 28442273 Free PMC article. Review.
-
Role of FXR in Bile Acid and Metabolic Homeostasis in NASH: Pathogenetic Concepts and Therapeutic Opportunities.Semin Liver Dis. 2021 Nov;41(4):461-475. doi: 10.1055/s-0041-1731707. Epub 2021 Jul 21. Semin Liver Dis. 2021. PMID: 34289507 Free PMC article. Review.
-
Bile acid receptors and nonalcoholic fatty liver disease.World J Hepatol. 2015 Dec 8;7(28):2811-8. doi: 10.4254/wjh.v7.i28.2811. World J Hepatol. 2015. PMID: 26668692 Free PMC article. Review.
-
Bile acid modulators for the treatment of nonalcoholic steatohepatitis (NASH).Expert Opin Investig Drugs. 2020 Jun;29(6):623-632. doi: 10.1080/13543784.2020.1763302. Epub 2020 Jun 19. Expert Opin Investig Drugs. 2020. PMID: 32552182 Review.
-
Interaction of gut microbiota with dysregulation of bile acids in the pathogenesis of nonalcoholic fatty liver disease and potential therapeutic implications of probiotics.J Cell Biochem. 2019 Mar;120(3):2713-2720. doi: 10.1002/jcb.27635. Epub 2018 Nov 15. J Cell Biochem. 2019. PMID: 30443932 Review.
Cited by
-
The role of the gut microbiota in health and cardiovascular diseases.Mol Biomed. 2022 Oct 11;3(1):30. doi: 10.1186/s43556-022-00091-2. Mol Biomed. 2022. PMID: 36219347 Free PMC article. Review.
-
Effects of Poria cocos extract on metabolic dysfunction-associated fatty liver disease via the FXR/PPARα-SREBPs pathway.Front Pharmacol. 2022 Oct 5;13:1007274. doi: 10.3389/fphar.2022.1007274. eCollection 2022. Front Pharmacol. 2022. PMID: 36278226 Free PMC article.
-
Interplay among IL1R1, gut microbiota, and bile acids in metabolic dysfunction-associated steatotic liver disease: a comprehensive review.J Gastroenterol Hepatol. 2025 Jan;40(1):33-40. doi: 10.1111/jgh.16750. Epub 2024 Sep 29. J Gastroenterol Hepatol. 2025. PMID: 39343617 Free PMC article. Review.
-
Unlocking therapeutic potential of amlexanox in MASH with insights into bile acid metabolism and microbiome.NPJ Gut Liver. 2025;2:4. doi: 10.1038/s44355-024-00015-7. Epub 2025 Feb 1. NPJ Gut Liver. 2025. PMID: 40519640 Free PMC article.
-
Utilization of Food-Derived β-Glucans to Prevent and Treat Non-Alcoholic Fatty Liver Disease (NAFLD).Foods. 2023 Sep 1;12(17):3279. doi: 10.3390/foods12173279. Foods. 2023. PMID: 37685211 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
