

The evolving story of apolipoprotein L1 nephropathy: the end of the beginning
- PMID: 35217848
- PMCID: PMC8877744
- DOI: 10.1038/s41581-022-00538-3
The evolving story of apolipoprotein L1 nephropathy: the end of the beginning
Abstract
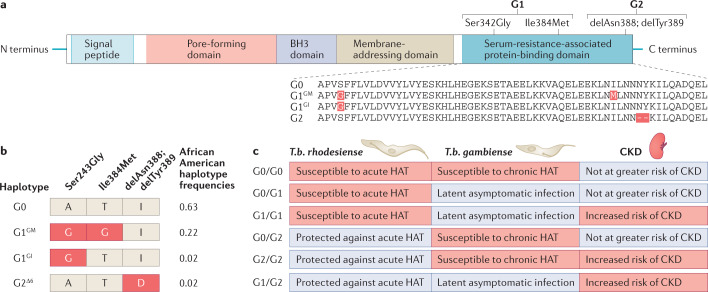
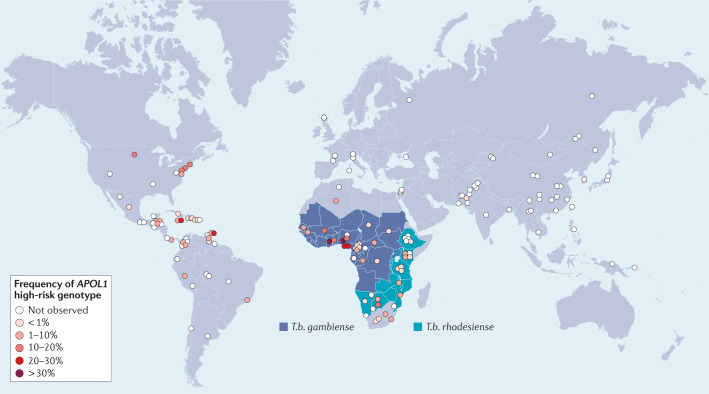
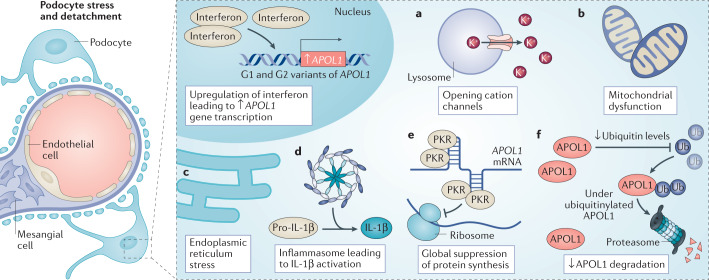
Genetic coding variants in APOL1, which encodes apolipoprotein L1 (APOL1), were identified in 2010 and are relatively common among individuals of sub-Saharan African ancestry. Approximately 13% of African Americans carry two APOL1 risk alleles. These variants, termed G1 and G2, are a frequent cause of kidney disease - termed APOL1 nephropathy - that typically manifests as focal segmental glomerulosclerosis and the clinical syndrome of hypertension and arterionephrosclerosis. Cell culture studies suggest that APOL1 variants cause cell dysfunction through several processes, including alterations in cation channel activity, inflammasome activation, increased endoplasmic reticulum stress, activation of protein kinase R, mitochondrial dysfunction and disruption of APOL1 ubiquitinylation. Risk of APOL1 nephropathy is mostly confined to individuals with two APOL1 risk variants. However, only a minority of individuals with two APOL1 risk alleles develop kidney disease, suggesting the need for a 'second hit'. The best recognized factor responsible for this 'second hit' is a chronic viral infection, particularly HIV-1, resulting in interferon-mediated activation of the APOL1 promoter, although most individuals with APOL1 nephropathy do not have an obvious cofactor. Current therapies for APOL1 nephropathies are not adequate to halt progression of chronic kidney disease, and new targeted molecular therapies are in clinical trials.
© 2022. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- The Churchill Society London. The Churchill Society http://www.churchill-society-london.org.uk/EndoBegn.html (1942).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
