

The β8 integrin cytoplasmic domain activates extracellular matrix adhesion to promote brain neurovascular development
- PMID: 35217866
- PMCID: PMC8977100
- DOI: 10.1242/dev.200472
The β8 integrin cytoplasmic domain activates extracellular matrix adhesion to promote brain neurovascular development
Abstract
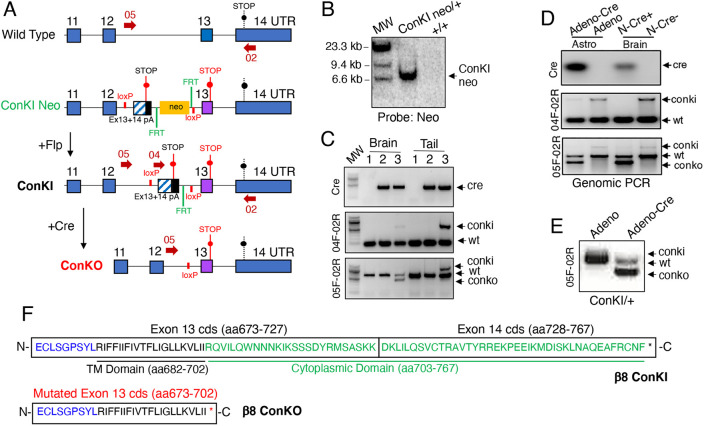
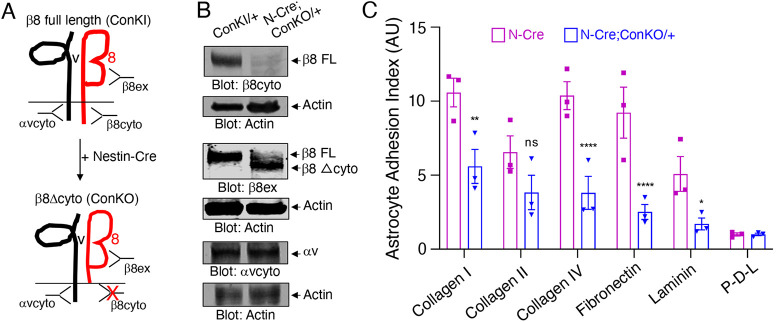
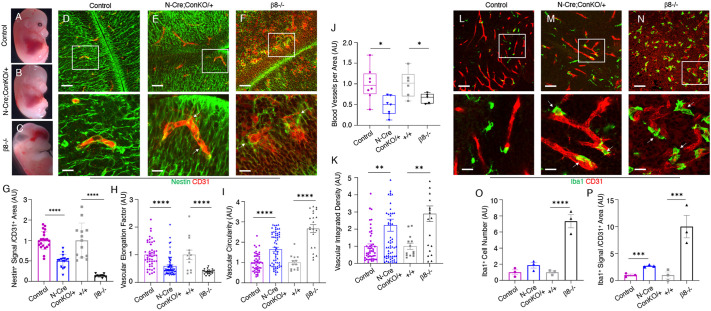
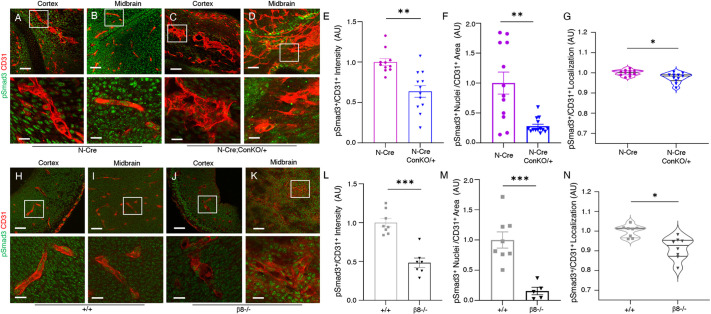
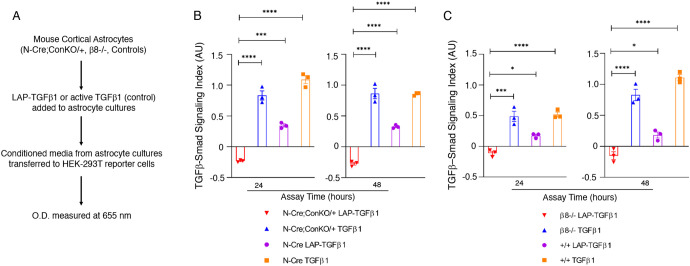
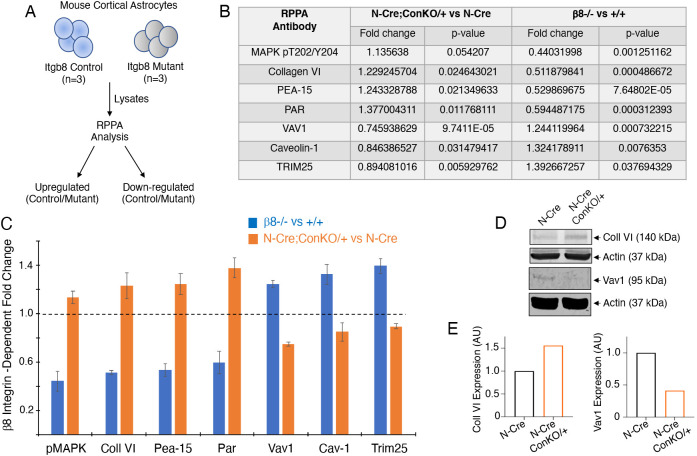
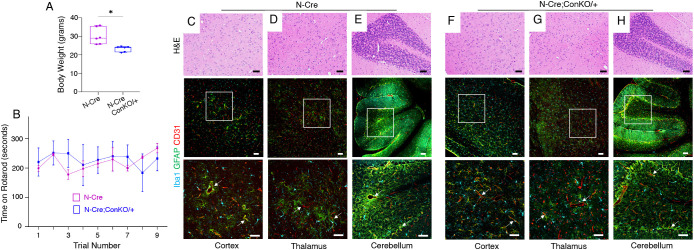
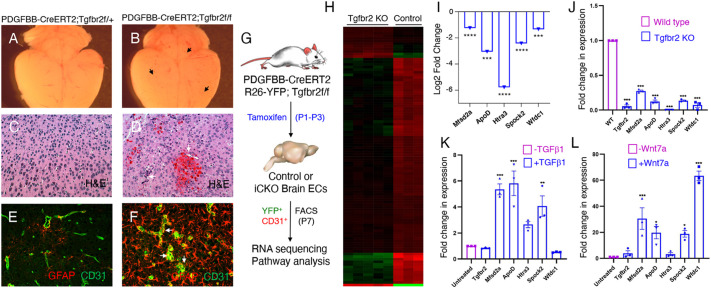
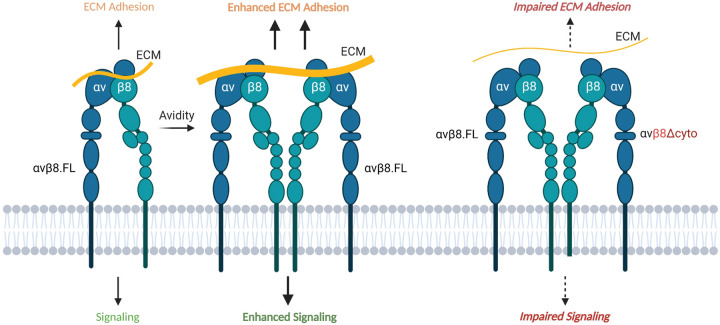
In the developing mammalian brain, neuroepithelial cells interact with blood vessels to regulate angiogenesis, blood-brain barrier maturation and other key neurovascular functions. Genetic studies in mice have shown that neurovascular development is controlled, in part, by Itgb8, which encodes the neuroepithelial cell-expressed integrin β8 subunit. However, these studies have involved complete loss-of-function Itgb8 mutations, and have not discerned the relative roles for the β8 integrin extracellular matrix (ECM) binding region versus the intracellular signaling tail. Here, Cre/lox strategies have been employed to selectively delete the cytoplasmic tail of murine Itgb8 without perturbing its transmembrane and extracellular domains. We report that the β8 integrin cytoplasmic domain is essential for inside-out modulation of adhesion, including activation of latent-TGFβs in the ECM. Quantitative sequencing of the brain endothelial cell transcriptome identifies TGFβ-regulated genes with putative links to blood vessel morphogenesis, including several genes linked to Wnt/β-catenin signaling. These results reveal that the β8 integrin cytoplasmic domain is essential for the regulation of TGFβ-dependent gene expression in endothelial cells and suggest that cross-talk between TGFβs and Wnt pathways is crucial for neurovascular development.
Keywords: Angiogenesis; Endothelial cell; Latent-TGFβ; Mouse; Neuroepithelial; Perivascular astrocyte; Wnt.
© 2022. Published by The Company of Biologists Ltd.
Conflict of interest statement
Competing interests The authors declare no competing or financial interests.
Figures
References
-
- Allinson, K. R., Lee, H. S., Fruttiger, M., McCarty, J. H. and Arthur, H. M. (2012). Endothelial expression of TGFβ type II receptor is required to maintain vascular integrity during postnatal development of the central nervous system. PLoS ONE 7, e39336. 10.1371/journal.pone.0039336 - DOI - PMC - PubMed
-
- Arnold, T. D., Ferrero, G. M., Qiu, H., Phan, I. T., Akhurst, R. J., Huang, E. J. and Reichardt, L. F. (2012). Defective retinal vascular endothelial cell development as a consequence of impaired integrin αVβ8-mediated activation of transforming growth factor-β. J. Neurosci. 32, 1197-1206. 10.1523/JNEUROSCI.5648-11.2012 - DOI - PMC - PubMed
-
- Arnold, T. D., Niaudet, C., Pang, M.-F., Siegenthaler, J., Gaengel, K., Jung, B., Ferrero, G. M., Mukouyama, Y.-S., Fuxe, J., Akhurst, R.et al. (2014). Excessive vascular sprouting underlies cerebral hemorrhage in mice lacking αVβ8-TGFβ signaling in the brain. Development 141, 4489-4499. 10.1242/dev.107193 - DOI - PMC - PubMed
-
- Arnold, T. D., Lizama, C. O., Cautivo, K. M., Santander, N., Lin, L., Qiu, H., Huang, E. J., Liu, C., Mukouyama, Y.-S., Reichardt, L. F.et al. (2019). Impaired alphaVbeta8 and TGFbeta signaling lead to microglial dysmaturation and neuromotor dysfunction. J. Exp. Med. 216, 900-915. 10.1084/jem.20181290 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
