

Y380Q novel mutation in receptor-binding domain of SARS-CoV-2 spike protein together with C379W interfere in the neutralizing antibodies interaction
- PMID: 35219552
- PMCID: PMC8761118
- DOI: 10.1016/j.diagmicrobio.2022.115636
Y380Q novel mutation in receptor-binding domain of SARS-CoV-2 spike protein together with C379W interfere in the neutralizing antibodies interaction
Abstract
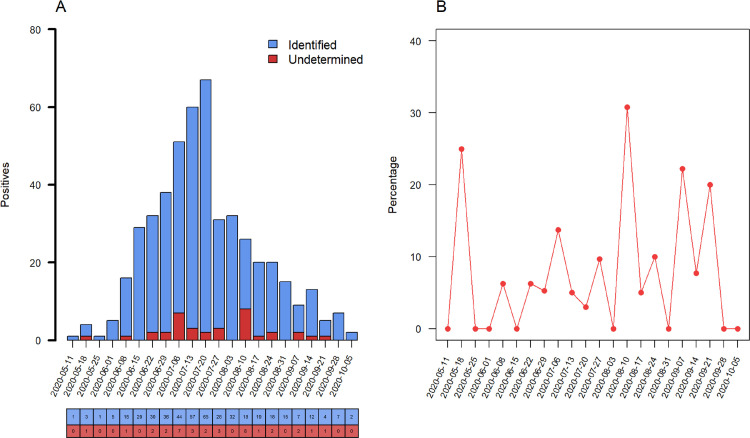
We aimed to describe the SARS-CoV-2 lineages circulating early pandemic among samples with S gene dropout and characterize the receptor-binding domain (RBD) of viral spike protein. Adults and children older than 2 months with signs and symptoms of COVID-19 were prospectively enrolled from May to October in Porto Alegre, Brazil. All participants performed RT-PCR assay, and samples with S gene dropout and cycle threshold < 30 were submitted to high-throughput sequencing (HTS). 484 out of 1,557 participants tested positive for SARS-CoV-2. The S gene dropout was detected in 7.4% (36/484) and a peak was observed in August. The B.1.1.28, B.1.91 and B.1.1.33 lineages were circulating in early pandemic. The RBD novel mutation (Y380Q) was found in one sample occurring simultaneously with C379W and V395A, and the B.1.91 lineage in the spike protein. The Y380Q and C379W may interfere with the binding of neutralizing antibodies (CR3022, EY6A, H014, S304).
Keywords: COVID-19; Novel mutation; RBD; SARS-CoV-2; Variants.
Copyright © 2022 Elsevier Inc. All rights reserved.
Figures
References
-
- cov-lineages/pangolin. CoV-lineages; 2021.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
