

Analysis of seed-associated bacteria and fungi on staple crops using the cultivation and metagenomic approaches
- PMID: 35220558
- PMCID: PMC9072454
- DOI: 10.1007/s12223-022-00958-5
Analysis of seed-associated bacteria and fungi on staple crops using the cultivation and metagenomic approaches
Abstract
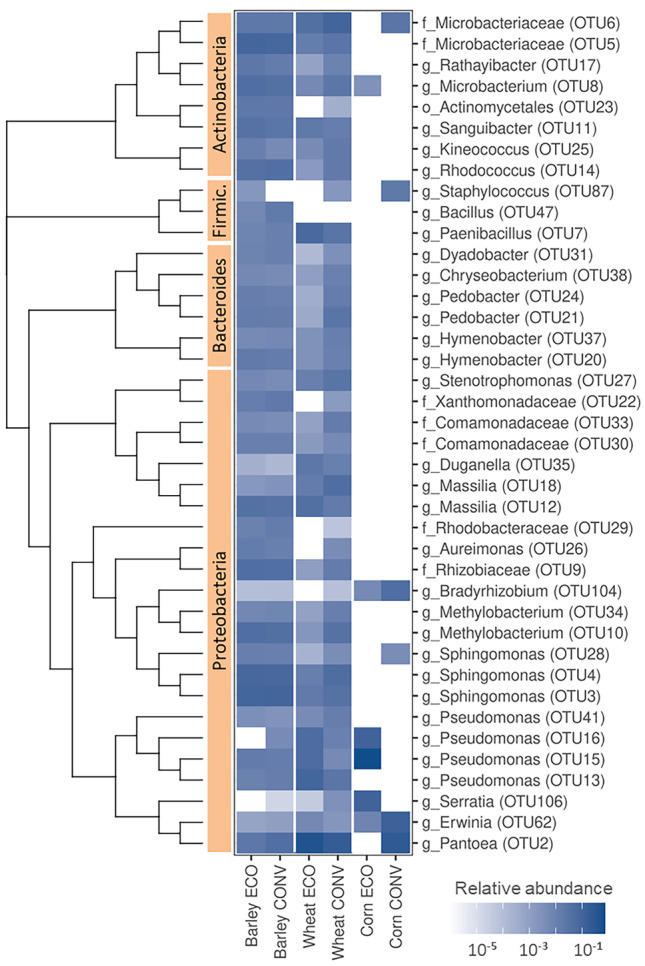
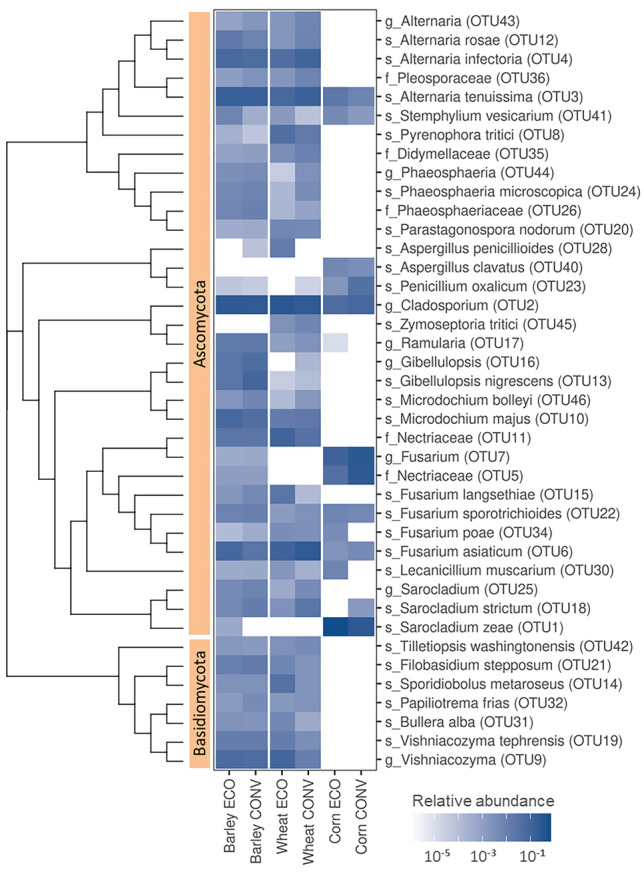
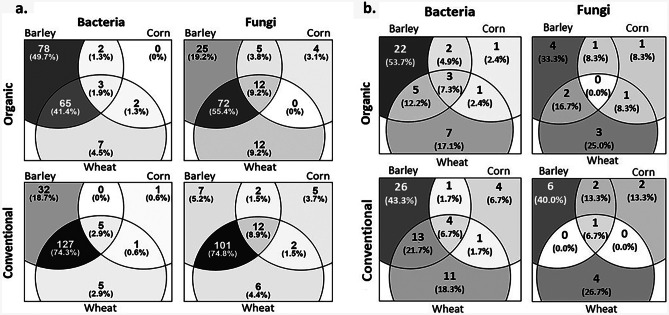
One of the key factors affecting seed quality is microbial communities residing on and in the seeds. In this study, microbial populations of seeds of conventionally and organically produced wheat, barley, and maize were analyzed using two different approaches: the cultivation method and metagenomics. For cultivation, three basic media were used: DG18 (for fungi), and nutrient agar or tryptic soy agar supplemented with cycloheximide or nystatin (for bacteria). Metagenomic sequencing was performed using the Illumina MiSeq platform. A total of 452 bacterial isolates comprising 36 genera and 5 phyla and 90 fungal isolates comprising 10 genera and 3 phyla were obtained from the seed surfaces. Among bacteria, representatives from the genera Bacillus, Pantoea, Paenibacillus, and Curtobacterium predominated, and among fungi, Aspergillus predominated. A total of 142 fungal OTUs and 201 bacterial OTUs were obtained from all the samples. Proteobacteria, Firmicutes, Bacteroides, and Actinobacteria comprised most of the bacterial OTUs, and Ascomycota comprised most of the fungal OTUs. Only 3 fungal OTUs (representatives of Curvibasidium, Venturia, and Dermateaceae) were exclusively present only within seeds and not on the seed surfaces. Barley seeds had the highest microbial load and richness, whereas corn had the lowest. Wheat and barley shared a higher number of OTUs than either of them did with corn with higher overlap between conventionally grown cereals than between organically grown cereals. Some OTUs were farming specific. This study demonstrates that the microbiome of cereal seeds is greatly dependent on the species of the host and is less affected by agricultural practices.
Keywords: Barley; Corn; Culture; Microbiota; NGS; Wheat.
© 2022. The Author(s).
Figures
Similar articles
-
Simultaneous profiling of seed-associated bacteria and fungi reveals antagonistic interactions between microorganisms within a shared epiphytic microbiome on Triticum and Brassica seeds.New Phytol. 2014 Apr;202(2):542-553. doi: 10.1111/nph.12693. Epub 2014 Jan 21. New Phytol. 2014. PMID: 24444052 Free PMC article.
-
Comparative Analysis of the Cultured and Total Bacterial Community in the Wheat Rhizosphere Microbiome Using Culture-Dependent and Culture-Independent Approaches.Microbiol Spectr. 2021 Oct 31;9(2):e0067821. doi: 10.1128/Spectrum.00678-21. Epub 2021 Oct 20. Microbiol Spectr. 2021. PMID: 34668733 Free PMC article.
-
Molecular Profiling on Surface-Disinfected Tomato Seeds Reveals High Diversity of Cultivation-Recalcitrant Endophytic Bacteria with Low Shares of Spore-Forming Firmicutes.Microb Ecol. 2020 May;79(4):910-924. doi: 10.1007/s00248-019-01440-5. Epub 2019 Nov 13. Microb Ecol. 2020. PMID: 31720799
-
The contribution of beneficial wheat seed fungal communities beyond disease-causing fungi: Advancing heritable mycobiome-based plant breeding.Environ Microbiol Rep. 2024 Dec;16(6):e70004. doi: 10.1111/1758-2229.70004. Environ Microbiol Rep. 2024. PMID: 39529232 Free PMC article. Review.
-
The seed microbiomes of staple food crops.Microb Biotechnol. 2023 Dec;16(12):2236-2249. doi: 10.1111/1751-7915.14352. Epub 2023 Oct 10. Microb Biotechnol. 2023. PMID: 37815330 Free PMC article. Review.
Cited by
-
Exploring Cereal Metagenomics: Unravelling Microbial Communities for Improved Food Security.Microorganisms. 2024 Mar 2;12(3):510. doi: 10.3390/microorganisms12030510. Microorganisms. 2024. PMID: 38543562 Free PMC article. Review.
-
Microbial inheritance through seed: a clouded area needs to be enlightened.Arch Microbiol. 2025 Jan 4;207(1):23. doi: 10.1007/s00203-024-04225-8. Arch Microbiol. 2025. PMID: 39754662 Review.
-
Community-forming traits play role in effective colonization of plant-growth-promoting bacteria and improved plant growth.Front Plant Sci. 2024 Mar 12;15:1332745. doi: 10.3389/fpls.2024.1332745. eCollection 2024. Front Plant Sci. 2024. PMID: 38533409 Free PMC article.
-
Vertical transfer and functional characterization of cotton seed core microbiome.Front Microbiol. 2024 Jan 9;14:1323342. doi: 10.3389/fmicb.2023.1323342. eCollection 2023. Front Microbiol. 2024. PMID: 38264479 Free PMC article.
-
Volatile Organic Compounds Produced by Kosakonia cowanii Cp1 Isolated from the Seeds of Capsicum pubescens R & P Possess Antifungal Activity.Microorganisms. 2023 Oct 4;11(10):2491. doi: 10.3390/microorganisms11102491. Microorganisms. 2023. PMID: 37894149 Free PMC article.
References
-
- Bengtsson-Palme J, Ryberg M, Hartmann M, Branco S, Wang Z, Godhe A, Wit P, Sánchez-García M, Ebersberger I, Sousa F, Amend A, Jumpponen A, Unterseher M, Kristiansson E, Abarenkov K, Bertrand Y, Sanli K, Eriksson KM, Vik U, Veldre V, Nilsson RH. Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol Evol. 2013;4:914–919. doi: 10.1111/2041-210X.12073. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
