

The Crohn Disease-associated ATG16L1T300A polymorphism regulates inflammatory responses by modulating TLR- and NLR-mediated signaling
- PMID: 35220902
- PMCID: PMC9629077
- DOI: 10.1080/15548627.2022.2039991
The Crohn Disease-associated ATG16L1T300A polymorphism regulates inflammatory responses by modulating TLR- and NLR-mediated signaling
Abstract
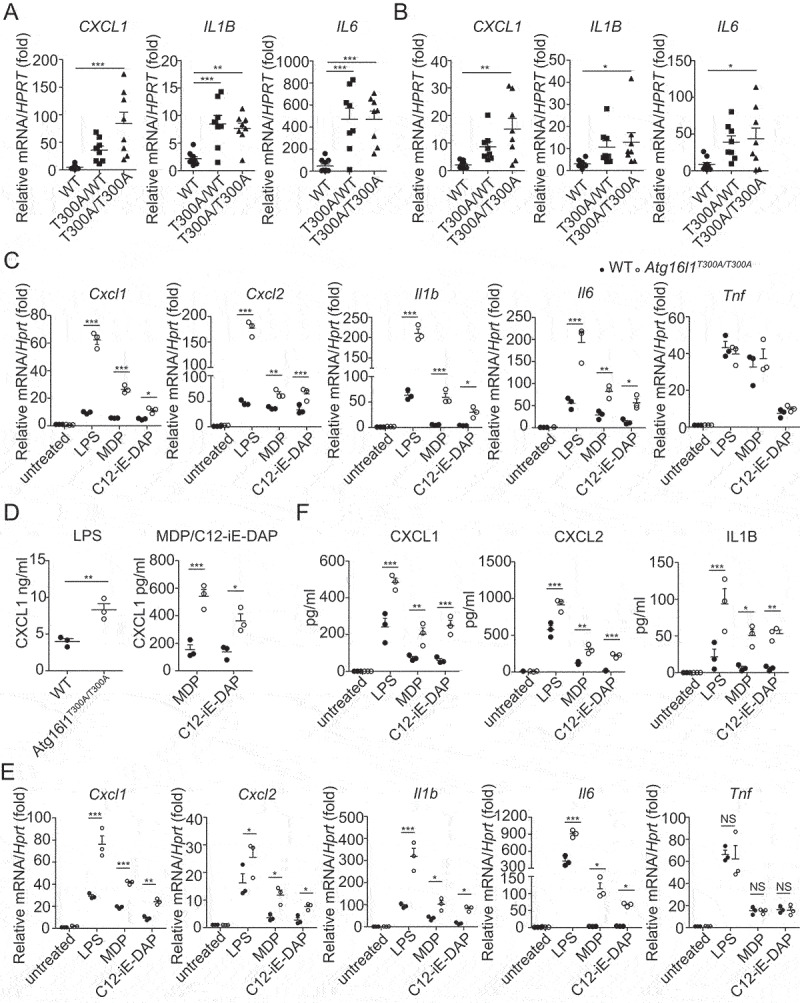
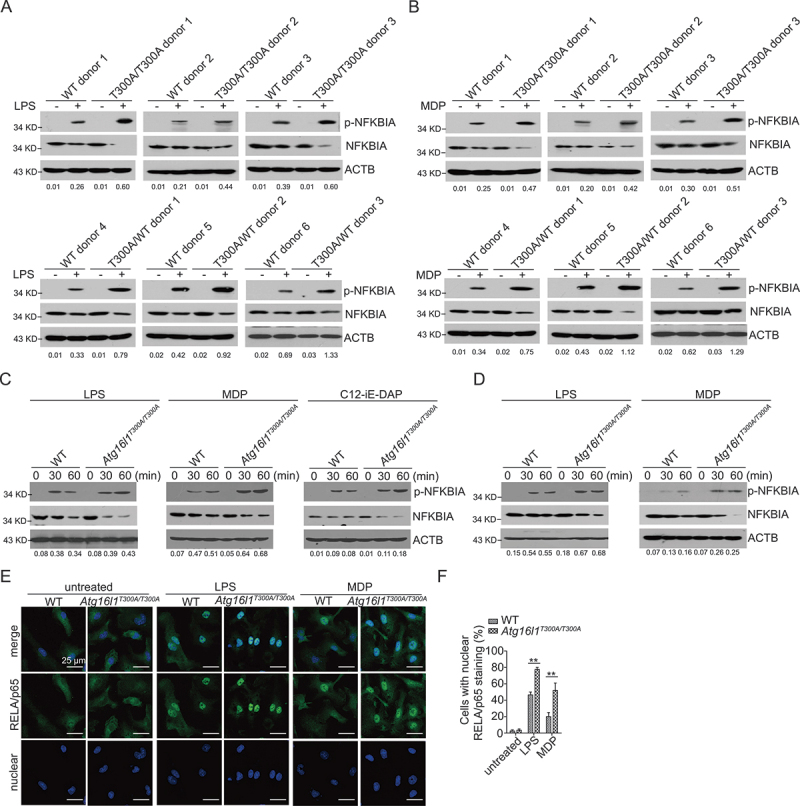
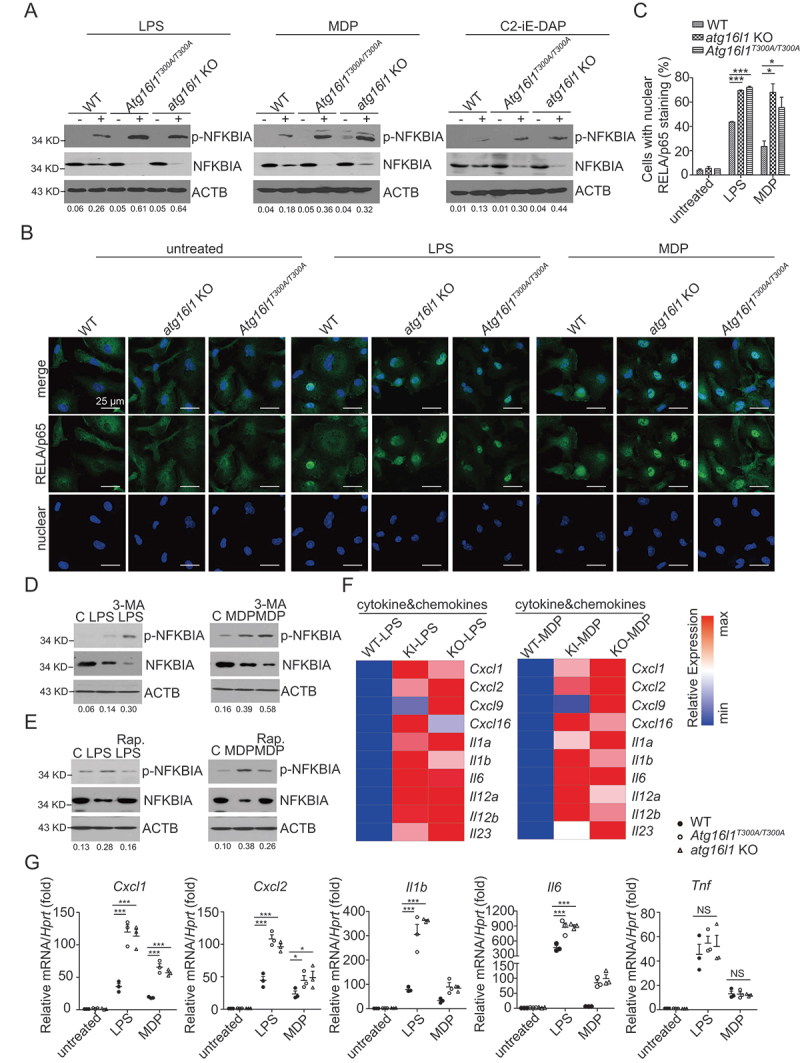
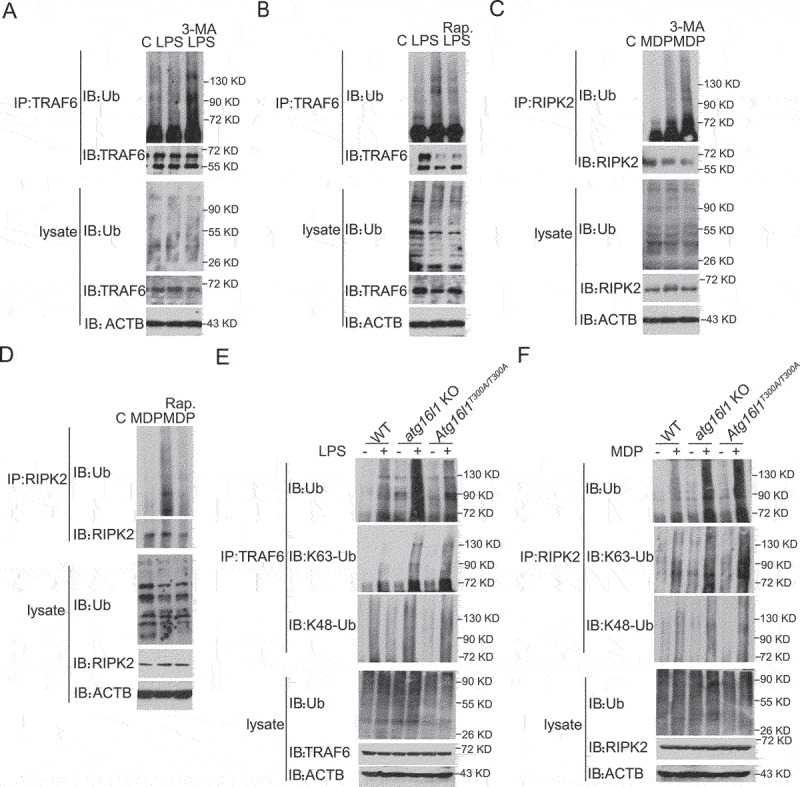
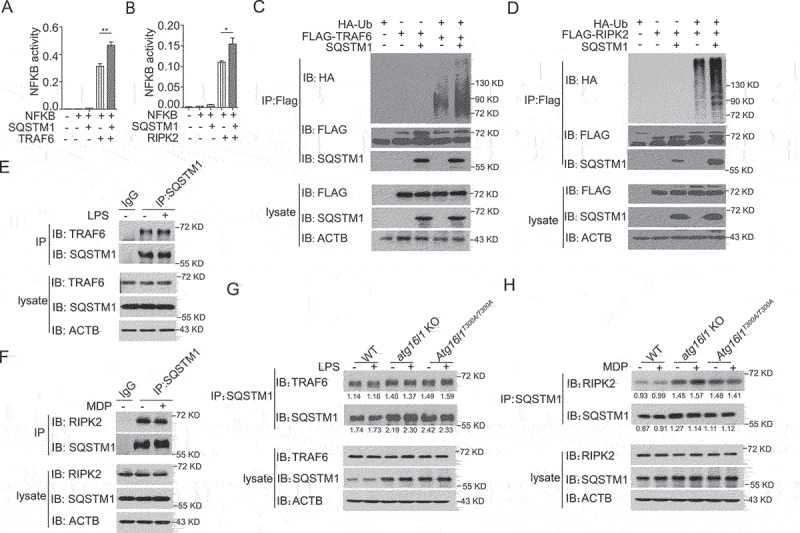
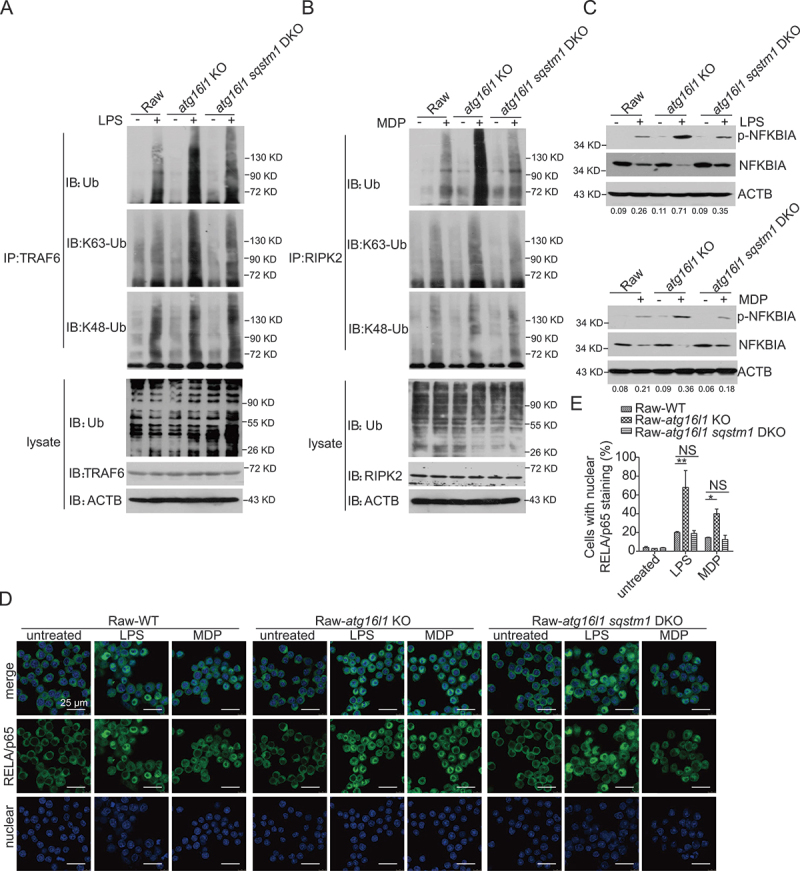
The mechanisms by which the ATG16L1T300A polymorphism affects cell function and causes an increased risk for the development of Crohn disease remain incompletely understood. Here we report that healthy individuals and mice bearing this polymorphism, even as heterozygotes, manifest enhanced TLR, and NLR cytokine and chemokine responses due to increased activation of NFKB. We elucidated the mechanism of the NFKB abnormality and found that in the ATG16L1T300A cell, there is enhanced polyubiquitination of TRAF6 or RIPK2 resulting from the accumulation of SQSTM1/p62. Indeed, knockout of Sqstm1 in autophagy-deficient cells almost completely normalized TRAF6 or RIPK2 polyubiquitination and NFKB activation in these cells. Thus, by identifying that autophagy is a pathway-intrinsic homeostatic mechanism that restricts excessive TLR- or NLR-mediated inflammatory signaling, our findings shed new light on how the ATG16L1T300A polymorphism sets the stage for the occurrence of Crohn disease.Abbreviations: 3-MA: 3-methyladenine; ATG16L1: autophagy related 16 like 1; ATG7: autophagy related 7; BMDM: bone marrow-derived macrophage; CD: Crohn disease; CXCL: C-X-C motif chemokine ligand; IBD: inflammatory bowel disease; iBMDM: immortalized mouse BMDM; IL1B/IL-1β: interleukin 1 beta; IL6: interleukin 6; KI: knockin; KO: knockout; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; LPS: lipopolysaccharide; MDP: muramyl dipeptide; MEF: mouse embryonic fibroblast; NFKB/NF-κB: nuclear factor kappa B; NFKBIA/IKBA: NFKB inhibitor alpha; NLR: NOD-like receptor; NOD: nucleotide-binding oligomerization domain containing; RIPK2: receptor interacting serine/threonine kinase 2; SNP: single nucleotide polymorphism; SQSTM1/p62: sequestosome 1; TLR: toll like receptor; TNF/TNF-α: tumor necrosis factor; TRAF6: TNF receptor associated factor 6; Ub: ubiquitin; WT: wild type.
Keywords: ATG16L1T300A; NFKB; NLR; TLR4; autophagy.
Conflict of interest statement
No potential conflict of interest was reported by the author(s).
Figures
References
-
- Hampe J, Franke A, Rosenstiel P, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39(2):207–211. - PubMed
-
- Murthy A, Li Y, Peng I, et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature. 2014;506(7489):456–462. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials