

Armored BCMA CAR T Cells Eliminate Multiple Myeloma and Are Resistant to the Suppressive Effects of TGF-β
- PMID: 35222421
- PMCID: PMC8863610
- DOI: 10.3389/fimmu.2022.832645
Armored BCMA CAR T Cells Eliminate Multiple Myeloma and Are Resistant to the Suppressive Effects of TGF-β
Abstract
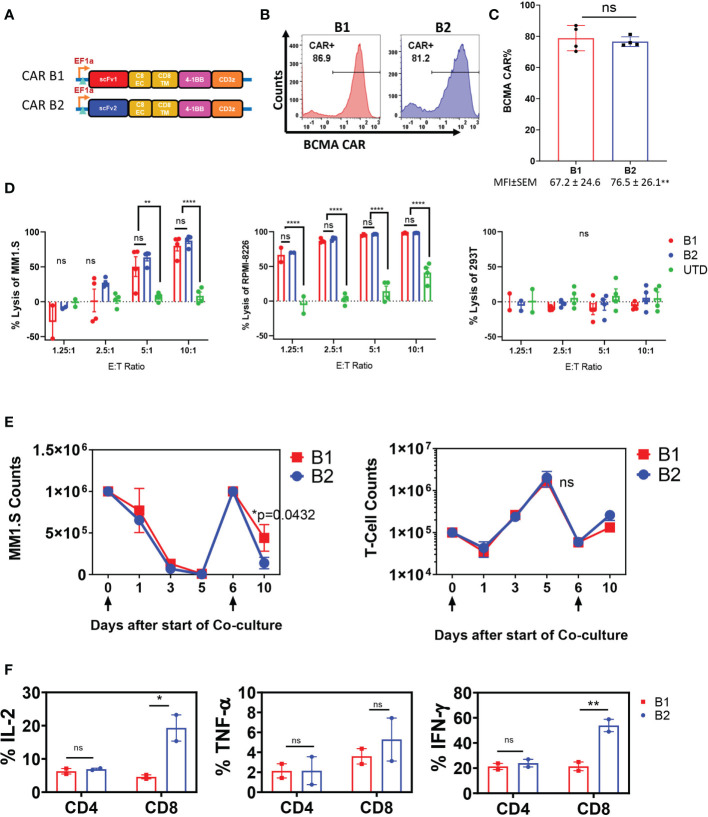
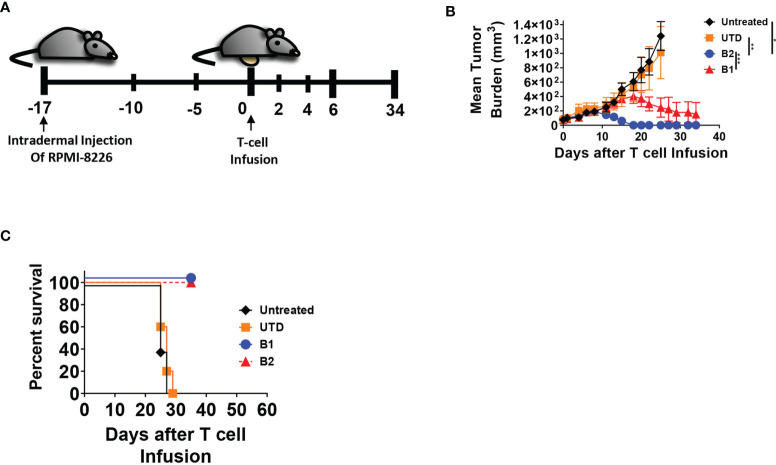
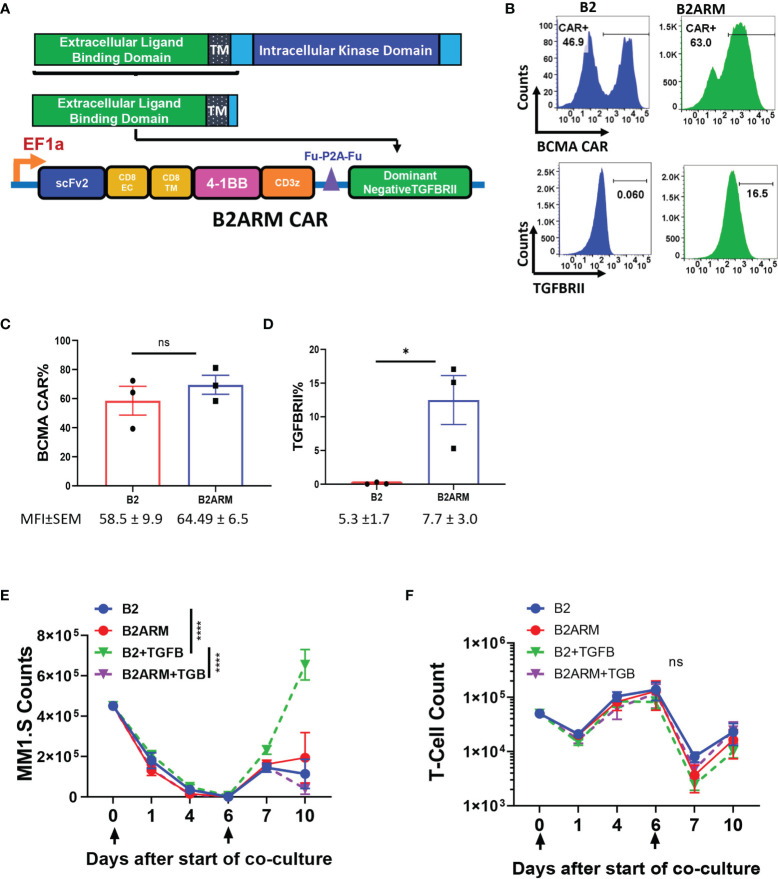
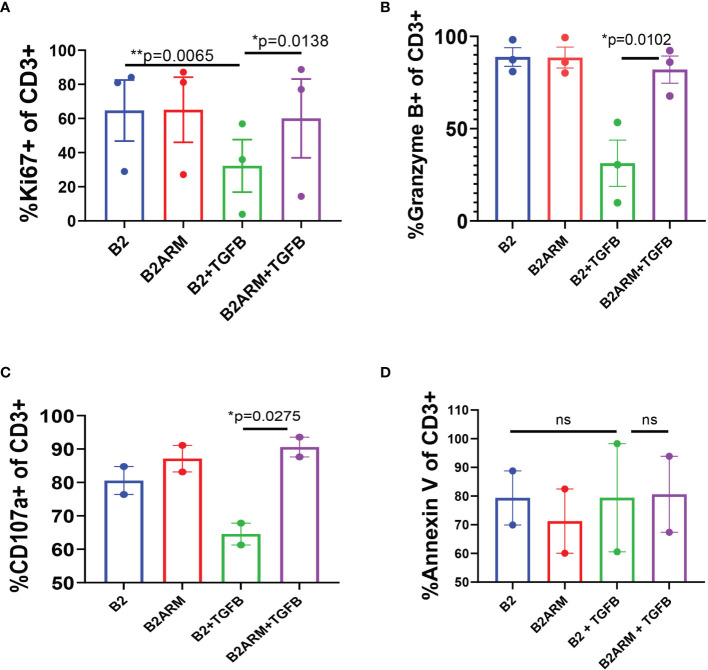
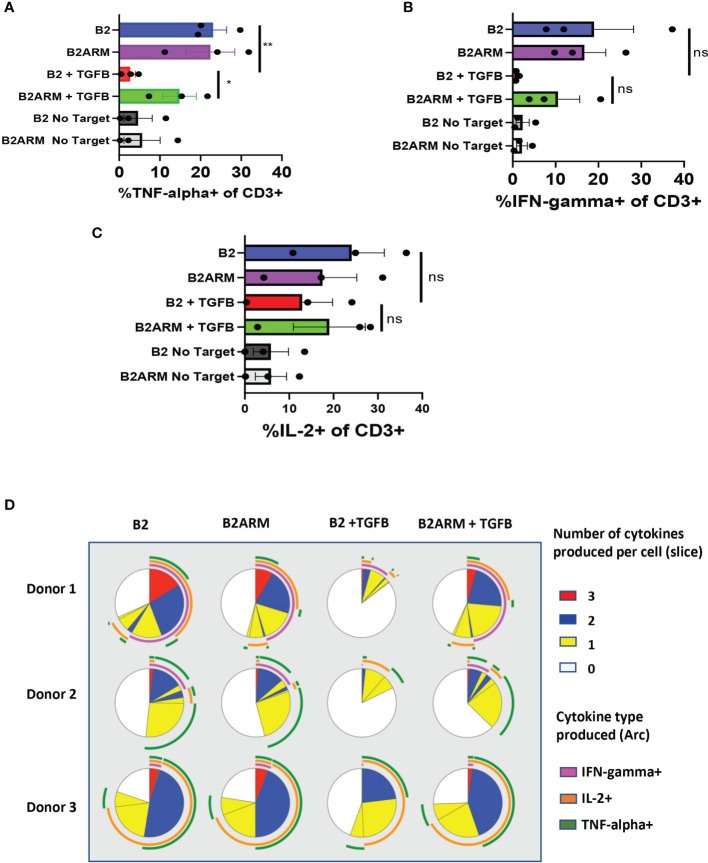
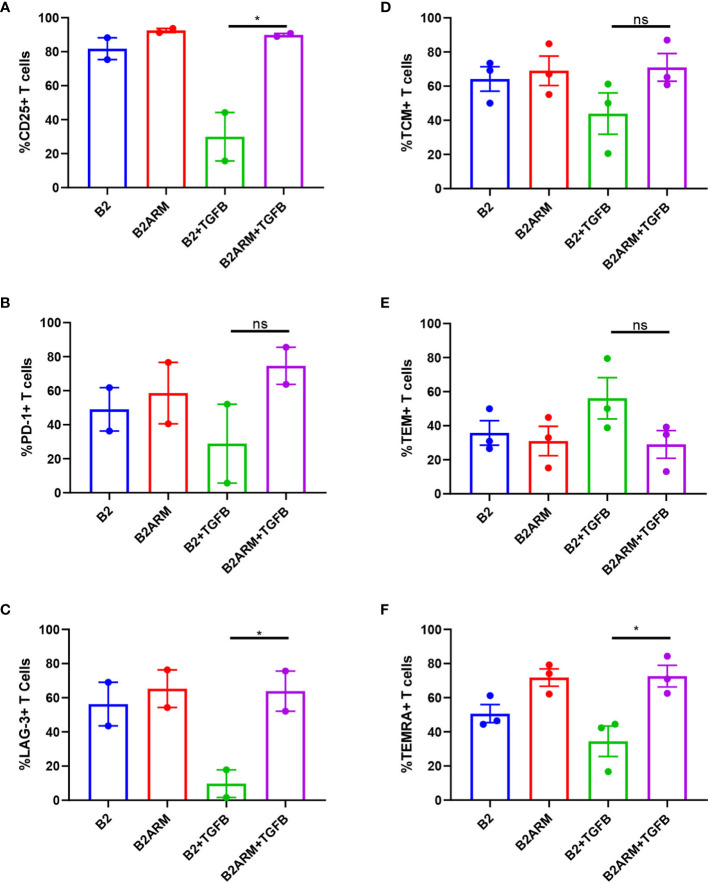
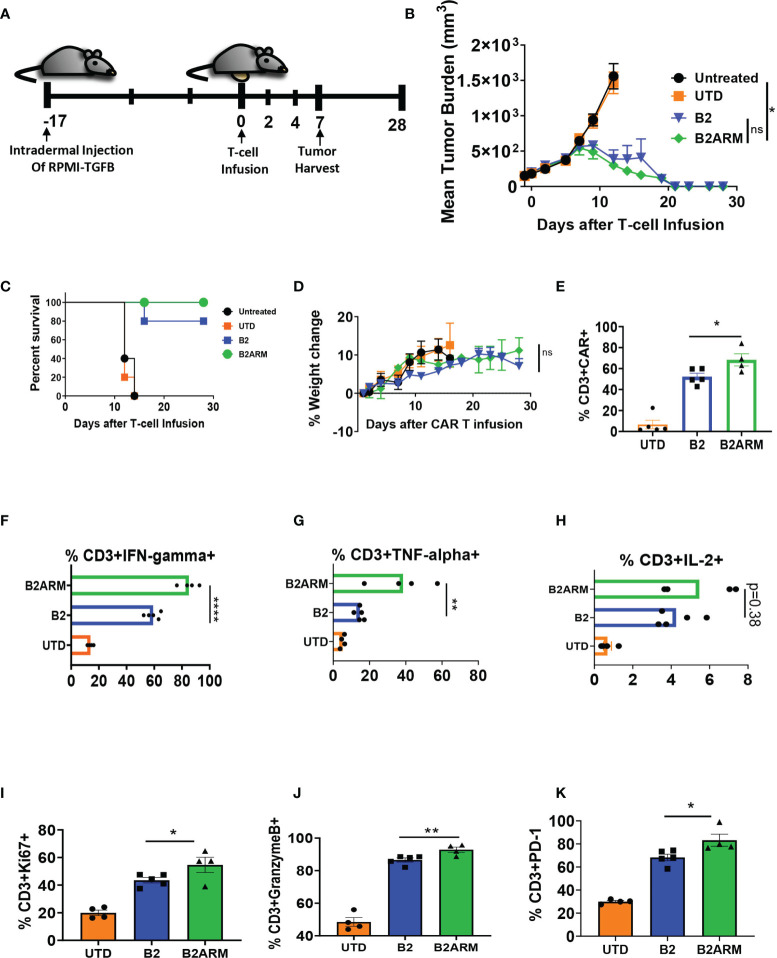
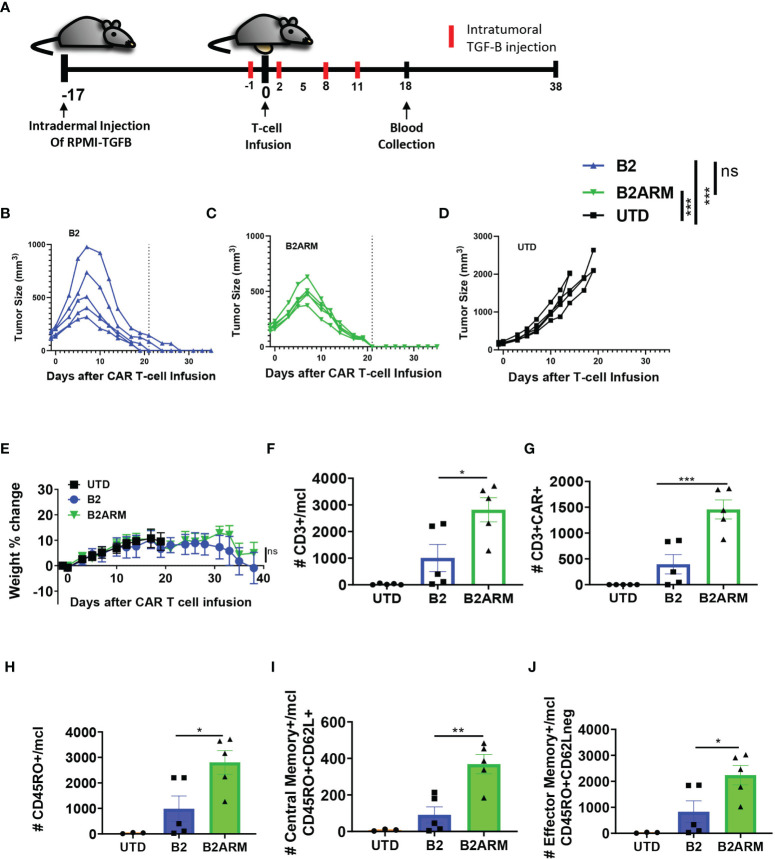
CAR T-cell therapies targeting the B-cell maturation antigen eliminate tumors in relapsed/refractory multiple myeloma patients, however durable remissions remain difficult to attain. Transforming growth factor beta (TGF-β) is a multifunctional cytokine abundantly expressed in the multiple myeloma bone marrow niche, where it promotes an immunosuppressive tumor microenvironment. We hypothesized that BCMA CAR T-cells armored to resist the suppressive effects of TGF-β will provide an advantage in treating multiple myeloma. The armored B2ARM CAR T cells, co-expressing BCMA targeting CAR with TGF-β dominant-negative receptor II, were generated by lentiviral transduction of primary human CD4+ and CD8+ T cells. The B2ARM CAR T cells eliminated MM.1S multiple myeloma targets in long-term cytotoxicity assays, even under TGF-β-high conditions, whereas cytotoxic function of the non-armored B2 CAR -T cells was inhibited by TGF-β. Concordantly, after long-term exposure to targets in the presence of TGF-β, the B2ARM CAR T cells were enriched for Granzyme B, CD107a, Ki67 and polyfunctional cells T-cells (double or triple-positive for IFN-γ, IL-2 and/or TNF-α), as determined by flow cytometry. In addition, the B2ARM CAR T-cells, but not the conventional B2 CAR T-cells, resisted the TGF-β-mediated suppression of activation (CD25), exhaustion (PD-1, LAG3), and differentiation to T effectors (CD45RA+ CD45RO-CD62L-). In NSG mice bearing RPMI-8226 tumors overexpressing TGF-β, the B2ARM CAR mediated 100% tumor rejection and survival, superior infiltration of tumors on day 7 post CAR T treatment (%CD3+CAR+), and greater expression of IFN-γ, TNF-α, Ki67, Granzyme B, and PD-1, as compared to tumor-infiltrating non-armored B2 CAR T-cells. In NSG RPMI-8226 xenograft model in which tumors were additionally supplemented with TGF-β injections on days -1 through 11 of CAR T treatment, the B2ARM CAR T cells rejected tumors faster than the non-armored B2 CARs, and showed greater numbers of CD3+ and CD3+CAR+, central memory (CD45RO+CD62L+) and effector memory (CD45RO+CD62L-) T cells in the peripheral blood 18 days after treatment. In summary, the armored B2ARM CAR T cells mediate superior persistence, proliferation, multi-functionality, effector differentiation and anti-tumor function in pre-clinical models of multiple myeloma, while abrogating TGF-β-mediated suppression.
Keywords: CAR T cells; TGF - β1; cell therapy; lentiviral (LV) vector; multiple myeloma.
Copyright © 2022 Alabanza, Xiong, Vu, Webster, Wu, Hu, Zhu, Dropulic, Dash and Schneider.
Conflict of interest statement
DS, LA, YX, ZZ, BV, DW, PH, PD are employees of Lentigen Technology, a Miltenyi Biotec Company. BW is an employee of Miltenyi Biotec. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Brudno JN, Maric I, Hartman SD, Rose JJ, Wang M, Lam N, et al. . T Cells Genetically Modified to Express an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J Clin Oncol (2018) 36(22):2267–80. doi: 10.1200/JCO.2018.77.8084 - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials