

Angiotensin II Induces Cardiac Edema and Hypertrophic Remodeling through Lymphatic-Dependent Mechanisms
- PMID: 35222798
- PMCID: PMC8881141
- DOI: 10.1155/2022/5044046
Angiotensin II Induces Cardiac Edema and Hypertrophic Remodeling through Lymphatic-Dependent Mechanisms
Abstract
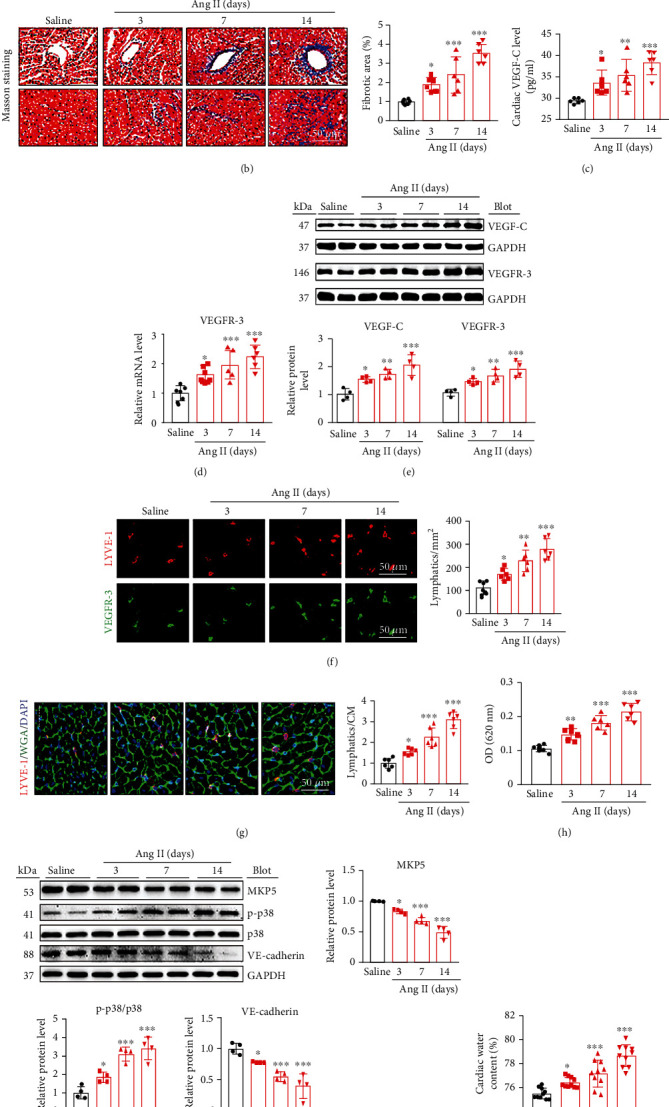
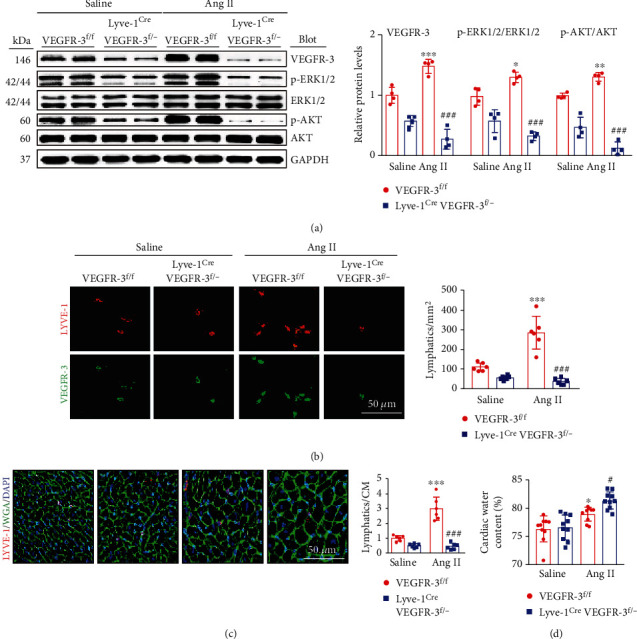
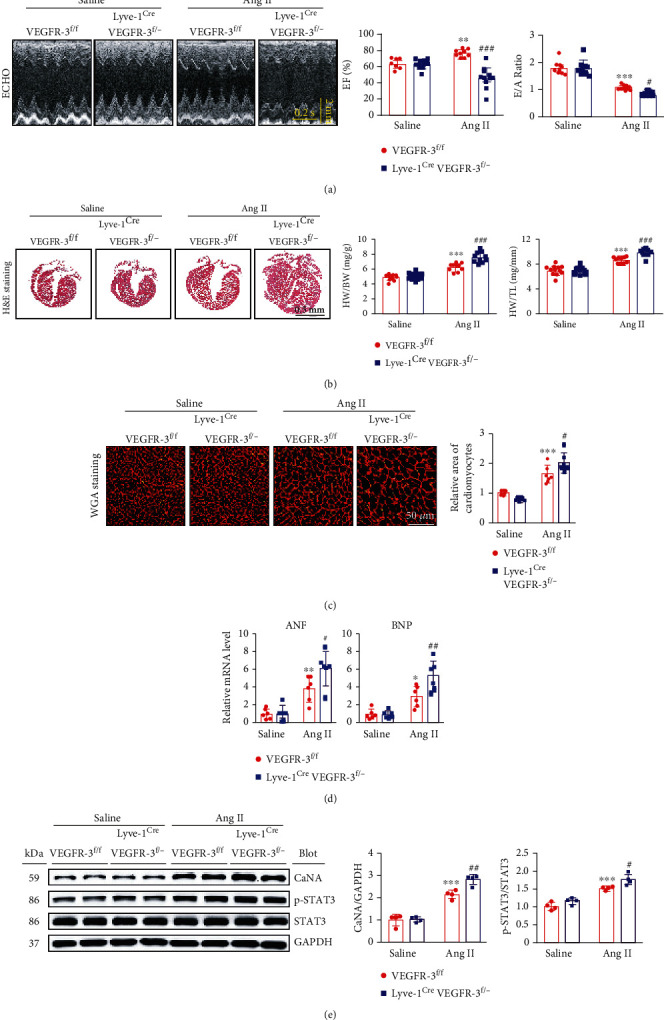
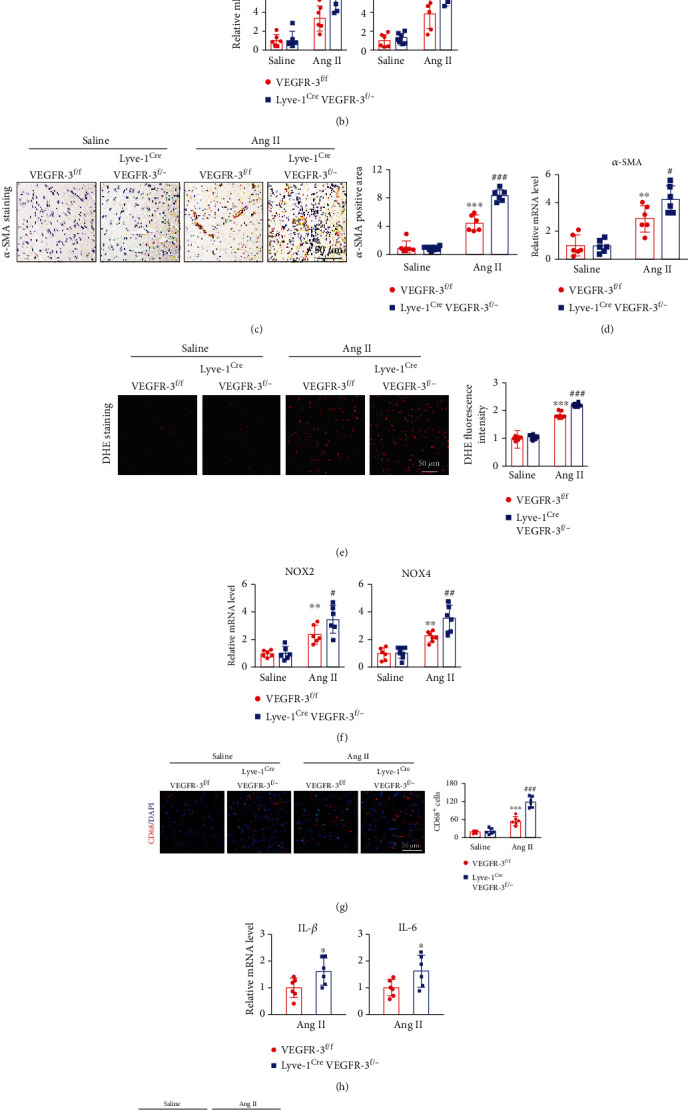
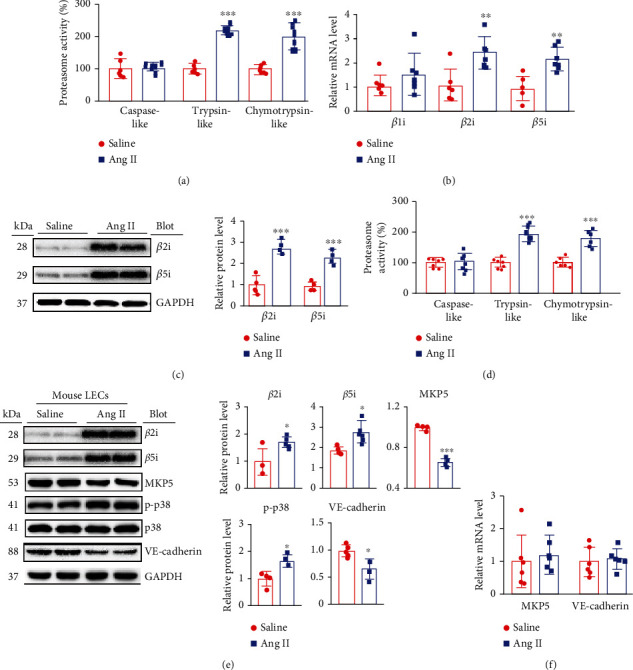
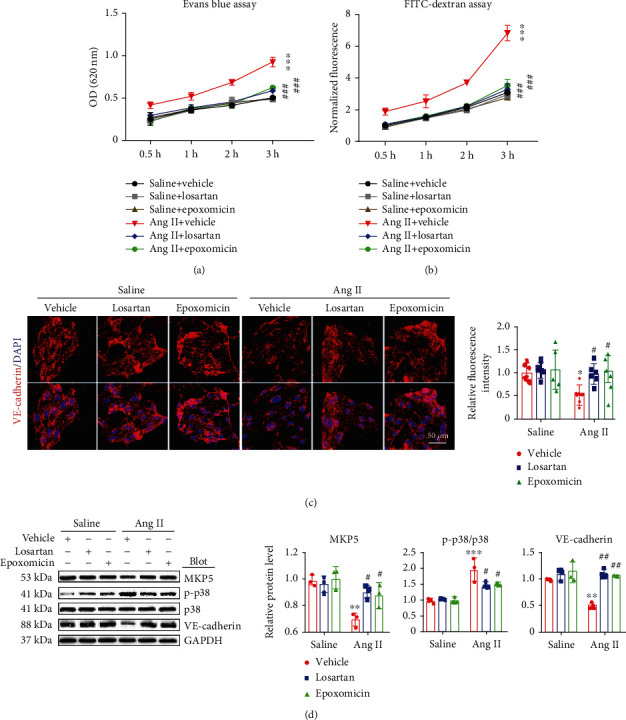
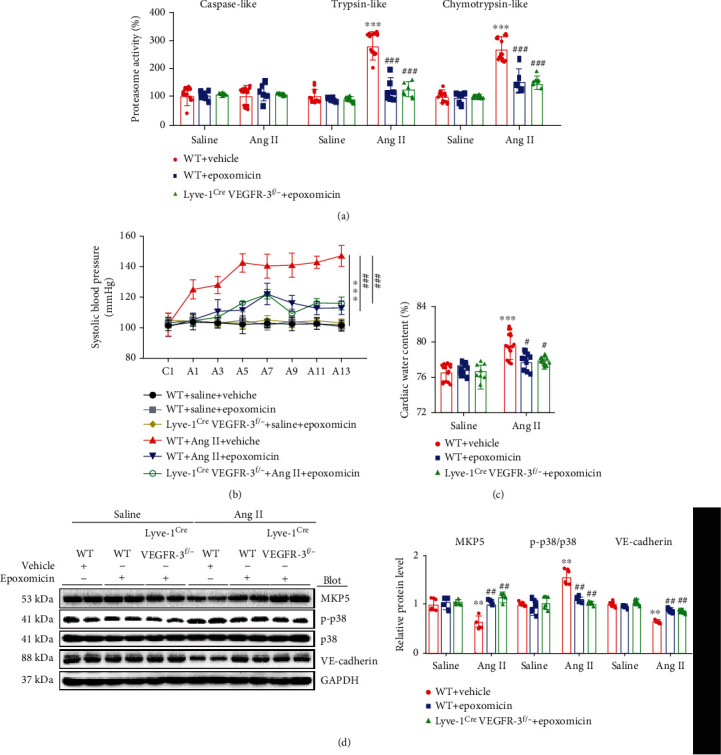
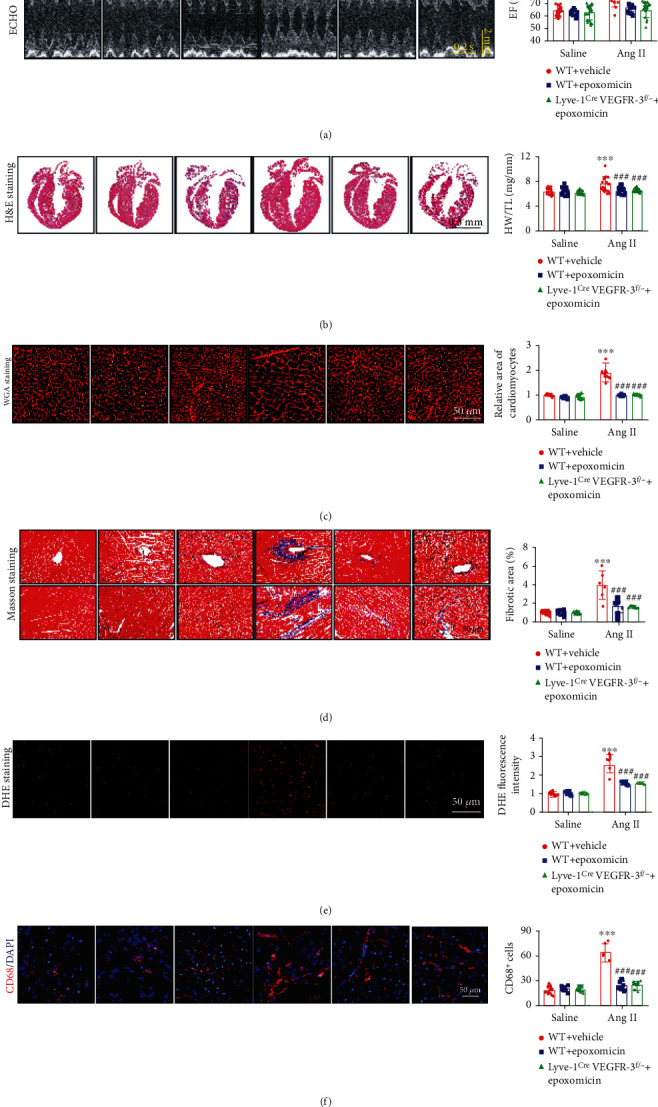
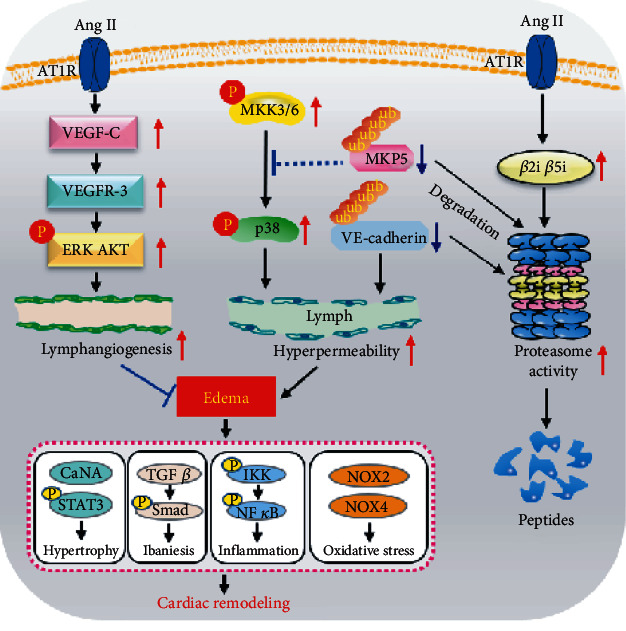
Cardiac lymphatic vessel growth (lymphangiogenesis) and integrity play an essential role in maintaining tissue fluid balance. Inhibition of lymphatic lymphangiogenesis is involved in cardiac edema and cardiac remodeling after ischemic injury or pressure overload. However, whether lymphatic vessel integrity is disrupted during angiotensin II- (Ang II-) induced cardiac remodeling remains to be investigated. In this study, cardiac remodeling models were established by Ang II (1000 ng/kg/min) in VEGFR-3 knockdown (Lyve-1Cre VEGFR-3f/-) and wild-type (VEGFR-3f/f) littermates. Our results indicated that Ang II infusion not only induced cardiac lymphangiogenesis and upregulation of VEGF-C and VEGFR-3 expression in the time-dependent manner but also enhanced proteasome activity, MKP5 and VE-cadherin degradation, p38 MAPK activation, and lymphatic vessel hyperpermeability. Moreover, VEGFR-3 knockdown significantly inhibited cardiac lymphangiogenesis in mice, resulting in exacerbation of tissue edema, hypertrophy, fibrosis superoxide production, inflammation, and heart failure (HF). Conversely, administration of epoxomicin (a selective proteasome inhibitor) markedly mitigated Ang II-induced cardiac edema, remodeling, and dysfunction; upregulated MKP5 and VE-cadherin expression; inactivated p38 MAPK; and reduced lymphatic vessel hyperpermeability in WT mice, indicating that inhibition of proteasome activity is required to maintain lymphatic endothelial cell (LEC) integrity. Our results show that both cardiac lymphangiogenesis and lymphatic barrier hyperpermeability are implicated in Ang II-induced adaptive hypertrophic remodeling and dysfunction. Proteasome-mediated hyperpermeability of LEC junctions plays a predominant role in the development of cardiac remodeling. Selective stimulation of lymphangiogenesis or inhibition of proteasome activity may be a potential therapeutic option for treating hypertension-induced cardiac remodeling.
Copyright © 2022 Jie Bai et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
