

Usher syndrome type IV: clinically and molecularly confirmed by novel ARSG variants
- PMID: 35226187
- PMCID: PMC9556359
- DOI: 10.1007/s00439-022-02441-0
Usher syndrome type IV: clinically and molecularly confirmed by novel ARSG variants
Abstract
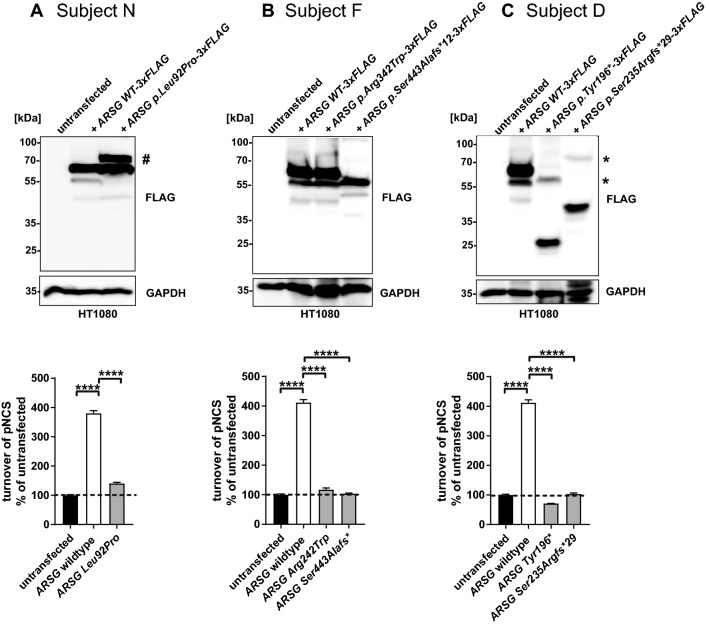
Usher syndrome (USH) is an autosomal recessively inherited disease characterized by sensorineural hearing loss (SNHL) and retinitis pigmentosa (RP) with or without vestibular dysfunction. It is highly heterogeneous both clinically and genetically. Recently, variants in the arylsulfatase G (ARSG) gene have been reported to underlie USH type IV. This distinct type of USH is characterized by late-onset RP with predominantly pericentral and macular changes, and late onset SNHL without vestibular dysfunction. In this study, we describe the USH type IV phenotype in three unrelated subjects. We identified three novel pathogenic variants, two novel likely pathogenic variants, and one previously described pathogenic variant in ARSG. Functional experiments indicated a loss of sulfatase activity of the mutant proteins. Our findings confirm that ARSG variants cause the newly defined USH type IV and support the proposed extension of the phenotypic USH classification.
© 2022. The Author(s).
Conflict of interest statement
The authors have no relevant financial or non-financial interests to disclose.
Figures



References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
