

Autoimmune Pulmonary Alveolar Proteinosis
- PMID: 35227171
- PMCID: PMC9851473
- DOI: 10.1164/rccm.202112-2742SO
Autoimmune Pulmonary Alveolar Proteinosis
Abstract
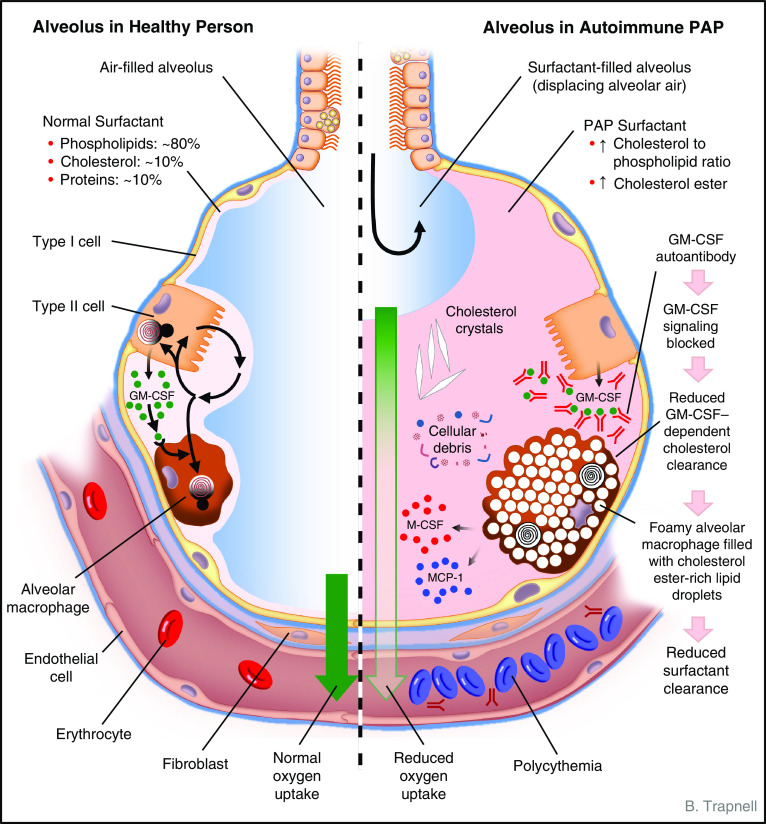
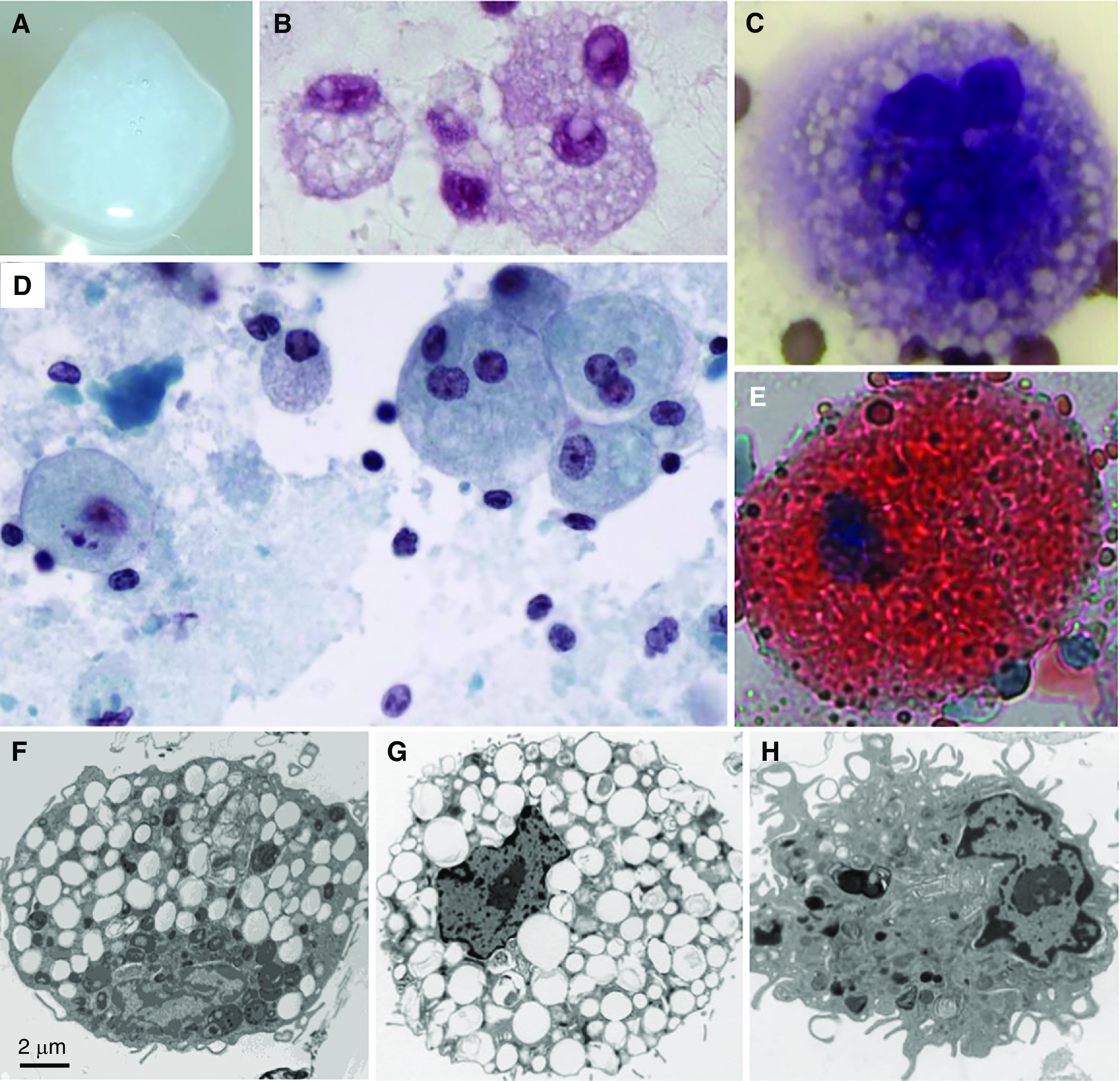
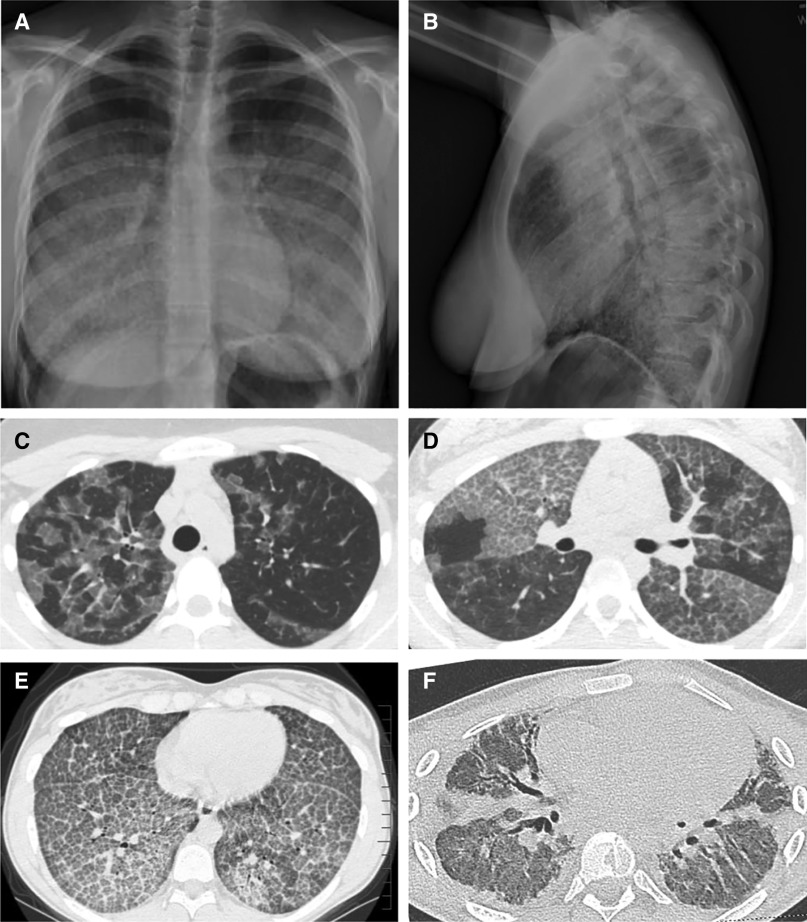
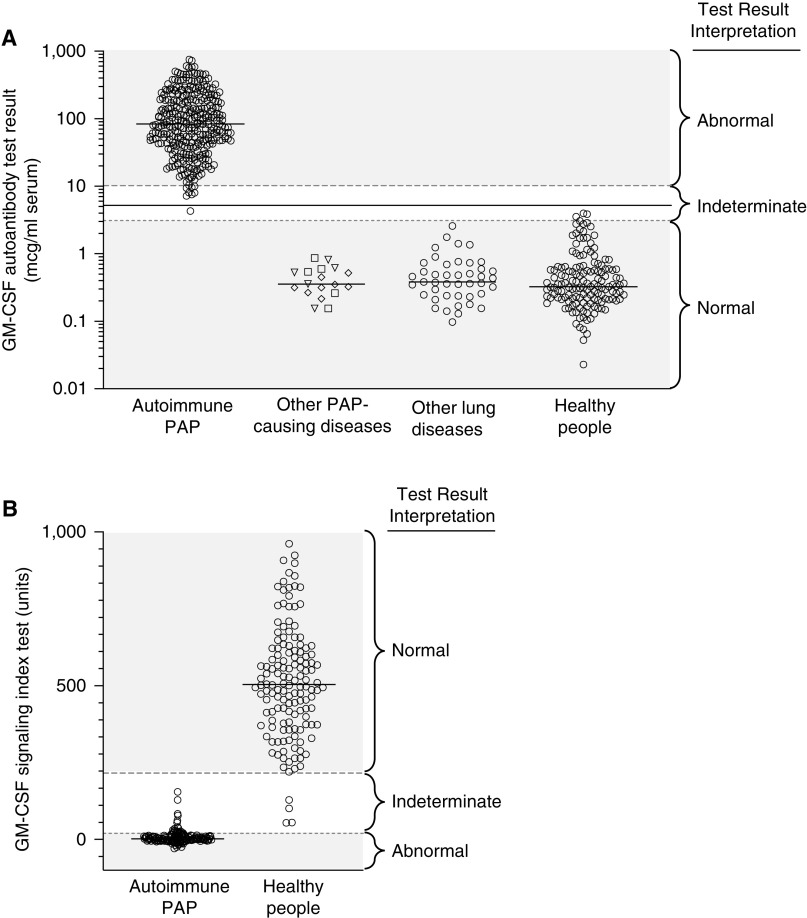
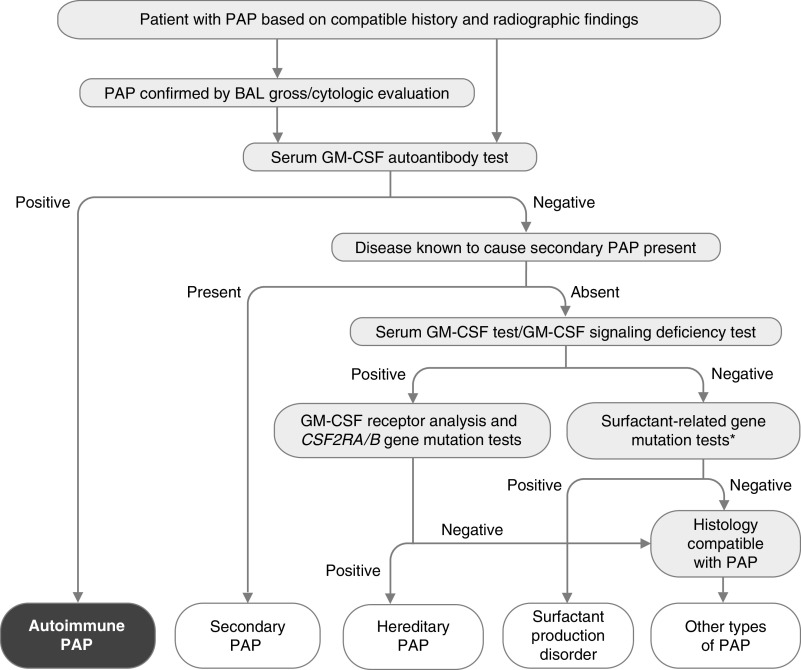
Autoimmune pulmonary alveolar proteinosis (PAP) is a rare disease characterized by myeloid cell dysfunction, abnormal pulmonary surfactant accumulation, and innate immune deficiency. It has a prevalence of 7-10 per million; occurs in individuals of all races, geographic regions, sex, and socioeconomic status; and accounts for 90% of all patients with PAP syndrome. The most common presentation is dyspnea of insidious onset with or without cough, production of scant white and frothy sputum, and diffuse radiographic infiltrates in a previously healthy adult, but it can also occur in children as young as 3 years. Digital clubbing, fever, and hemoptysis are not typical, and the latter two indicate that intercurrent infection may be present. Low prevalence and nonspecific clinical, radiological, and laboratory findings commonly lead to misdiagnosis as pneumonia and substantially delay an accurate diagnosis. The clinical course, although variable, usually includes progressive hypoxemic respiratory insufficiency and, in some patients, secondary infections, pulmonary fibrosis, respiratory failure, and death. Two decades of research have raised autoimmune PAP from obscurity to a paradigm of molecular pathogenesis-based diagnostic and therapeutic development. Pathogenesis is driven by GM-CSF (granulocyte/macrophage colony-stimulating factor) autoantibodies, which are present at high concentrations in blood and tissues and form the basis of an accurate, commercially available diagnostic blood test with sensitivity and specificity of 100%. Although whole-lung lavage remains the first-line therapy, inhaled GM-CSF is a promising pharmacotherapeutic approach demonstrated in well-controlled trials to be safe, well tolerated, and efficacious. Research has established GM-CSF as a pulmonary regulatory molecule critical to surfactant homeostasis, alveolar stability, lung function, and host defense.
Keywords: BAL; alveolar macrophages; autoantibodies; granulocyte/macrophage colony–stimulating factor; surfactant.
Figures
References
-
- Rosen SH, Castleman B, Liebow AA. Pulmonary alveolar proteinosis. N Engl J Med . 1958;258:1123–1142. - PubMed
-
- Trapnell BC, Whitsett JA, Nakata K. Pulmonary alveolar proteinosis. N Engl J Med . 2003;349:2527–2539. - PubMed
-
- Seymour JF, Presneill JJ. Pulmonary alveolar proteinosis: progress in the first 44 years. Am J Respir Crit Care Med . 2002;166:215–235. - PubMed
-
- Trapnell BC, Nakata K, Bonella F, Campo I, Griese M, Hamilton J, et al. Pulmonary alveolar proteinosis. Nat Rev Dis Primers . 2019;5:16. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
