

Single-cell gene fusion detection by scFusion
- PMID: 35228538
- PMCID: PMC8885711
- DOI: 10.1038/s41467-022-28661-6
Single-cell gene fusion detection by scFusion
Abstract
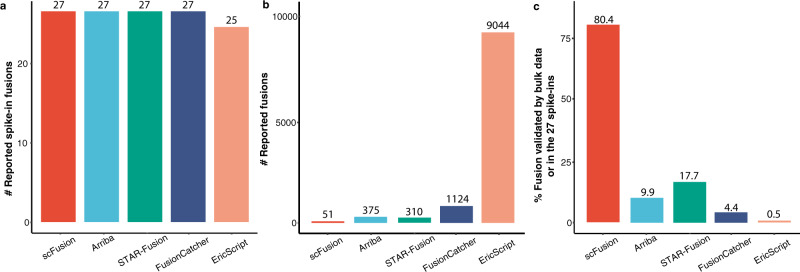
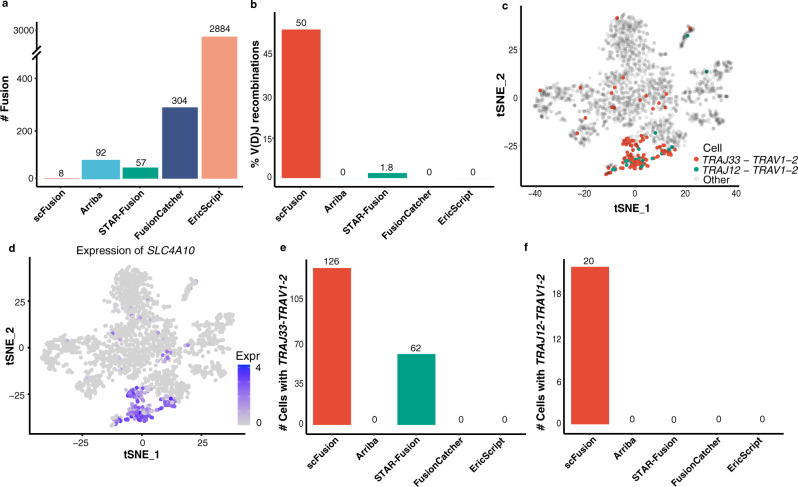
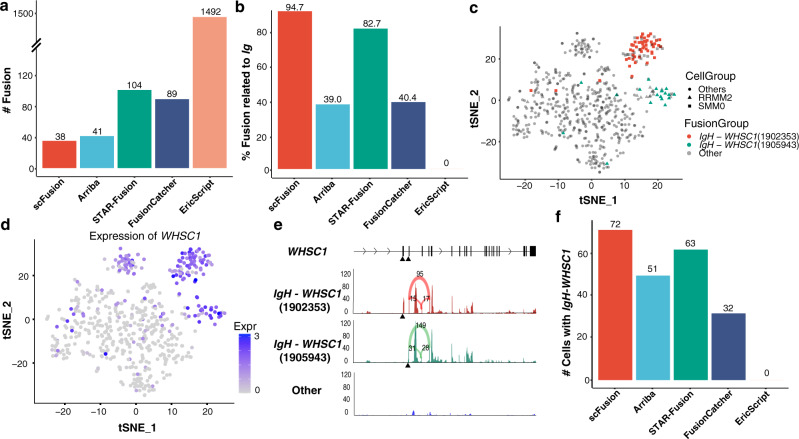
Gene fusions can play important roles in tumor initiation and progression. While fusion detection so far has been from bulk samples, full-length single-cell RNA sequencing (scRNA-seq) offers the possibility of detecting gene fusions at the single-cell level. However, scRNA-seq data have a high noise level and contain various technical artifacts that can lead to spurious fusion discoveries. Here, we present a computational tool, scFusion, for gene fusion detection based on scRNA-seq. We evaluate the performance of scFusion using simulated and five real scRNA-seq datasets and find that scFusion can efficiently and sensitively detect fusions with a low false discovery rate. In a T cell dataset, scFusion detects the invariant TCR gene recombinations in mucosal-associated invariant T cells that many methods developed for bulk data fail to detect; in a multiple myeloma dataset, scFusion detects the known recurrent fusion IgH-WHSC1, which is associated with overexpression of the WHSC1 oncogene. Our results demonstrate that scFusion can be used to investigate cellular heterogeneity of gene fusions and their transcriptional impact at the single-cell level.
© 2022. The Author(s).
Conflict of interest statement
The authors declare the following competing interests: Ruibin Xi holds the stock of GeneX Health Co.Ltd. A patent application about single-cell gene fusion detection is submitted. Applicant: Peking University. Inventors: Ruibin Xi, Zijie Jin. Application number: 202011451710.8. Status of the application: pending. The algorithm developed in this manuscript is covered in the patent application. For all other authors, no competing interests exist.
Figures
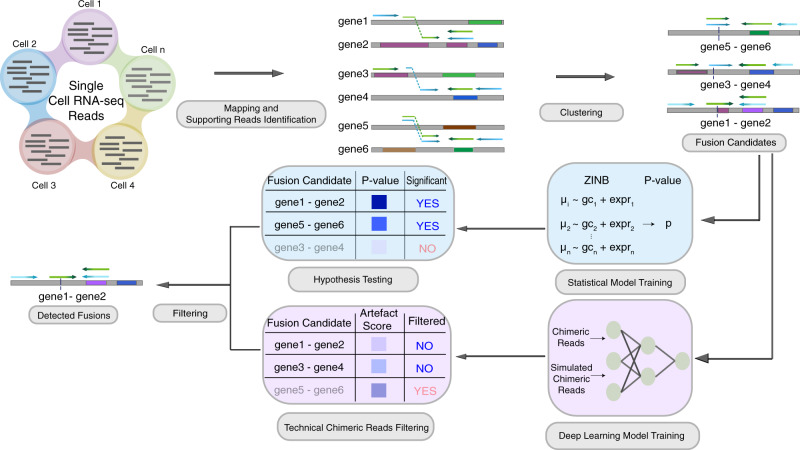
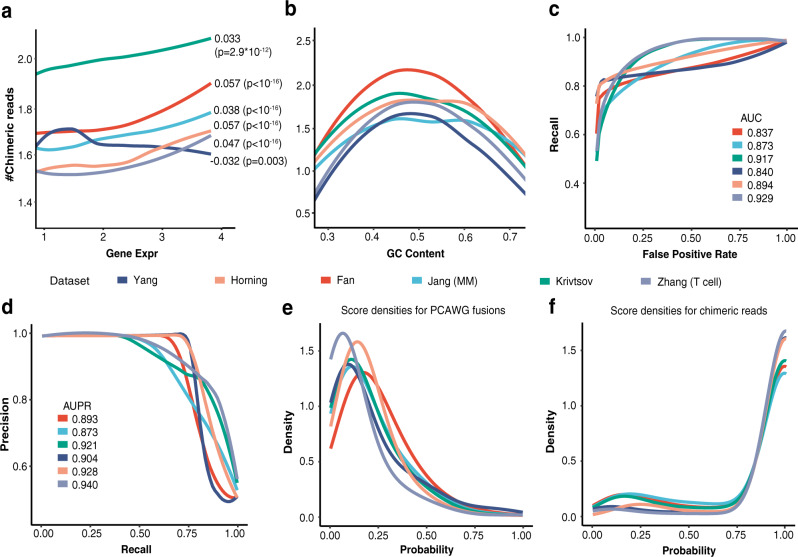
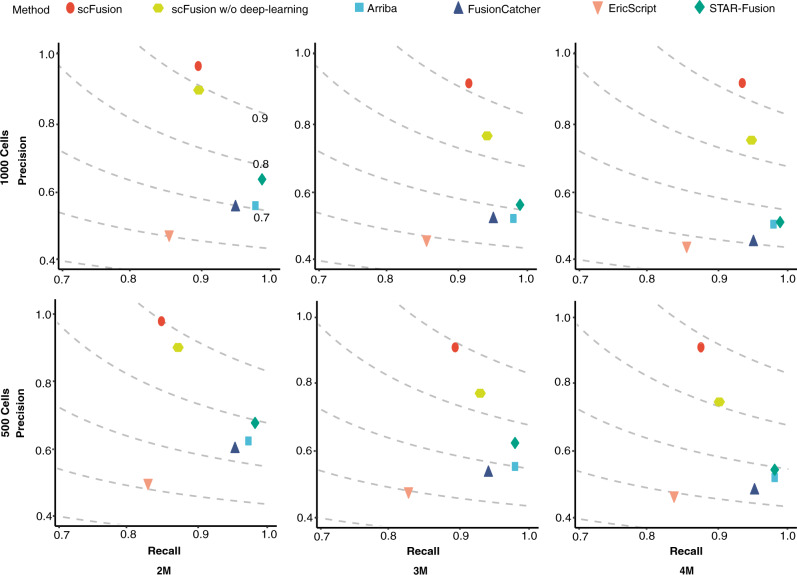
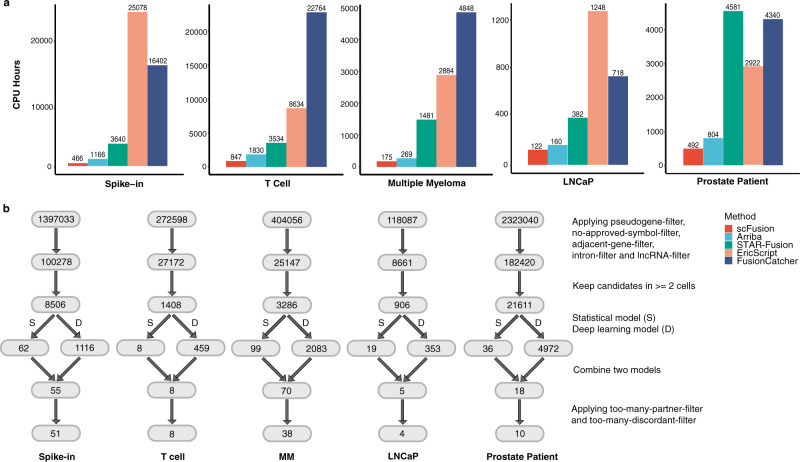
References
-
- Rowley JD. Identificaton of a translocation with quinacrine fluorescence in a patient with acute leukemia. Annal. Genetique. 1973;16:109–112. - PubMed
-
- Nowell PC, Hungerford DA. Chromosome studies on normal and leukemic human leukocytes. J. Natl Cancer Inst. 1960;25:85–109. - PubMed
-
- Demichelis F, et al. TMPRSS2:ERG gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene. 2007;26:4596–4599. - PubMed
-
- Choi YL, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N. Engl. J. Med. 2010;363:1734–1739. - PubMed
-
- O’Hare T, et al. In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res. 2005;65:4500–4505. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
