

Cross-cohort gut microbiome associations with immune checkpoint inhibitor response in advanced melanoma
- PMID: 35228751
- PMCID: PMC8938272
- DOI: 10.1038/s41591-022-01695-5
Cross-cohort gut microbiome associations with immune checkpoint inhibitor response in advanced melanoma
Abstract
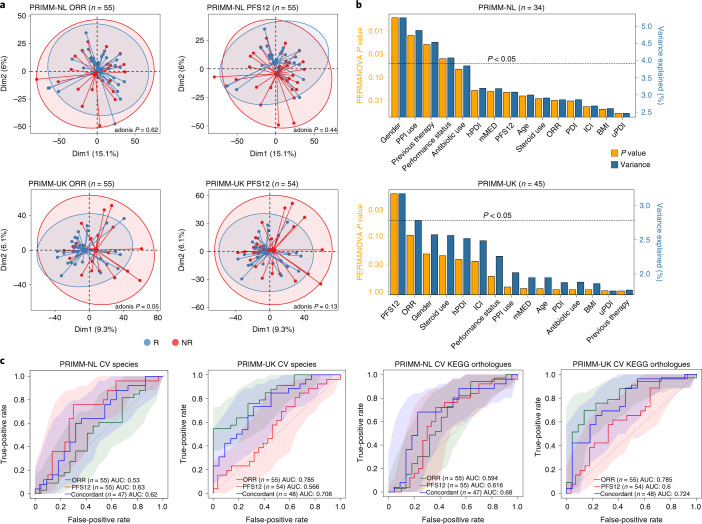
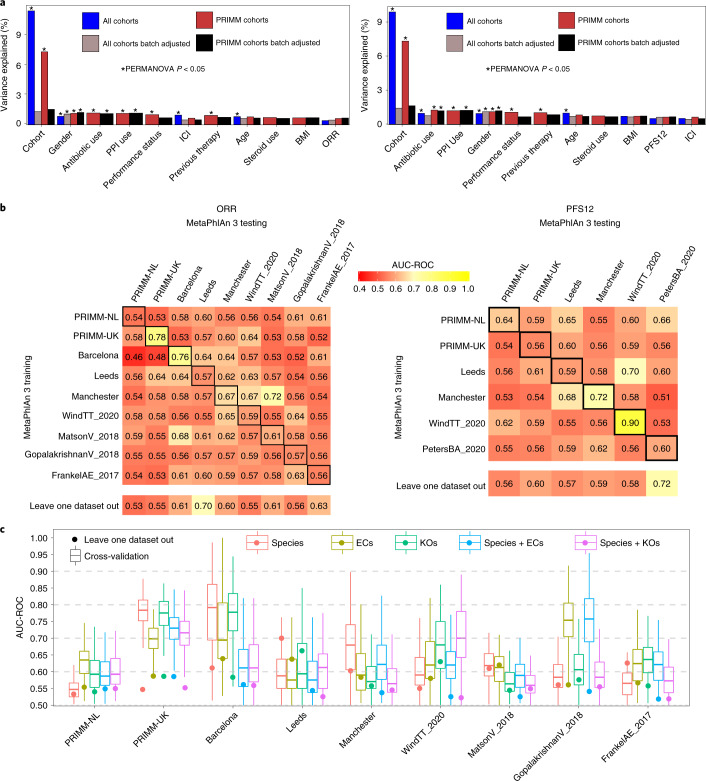
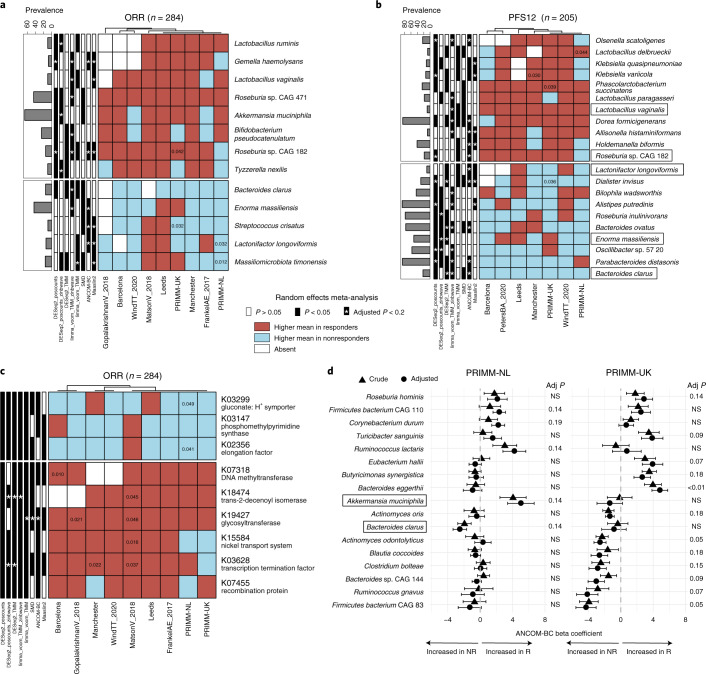
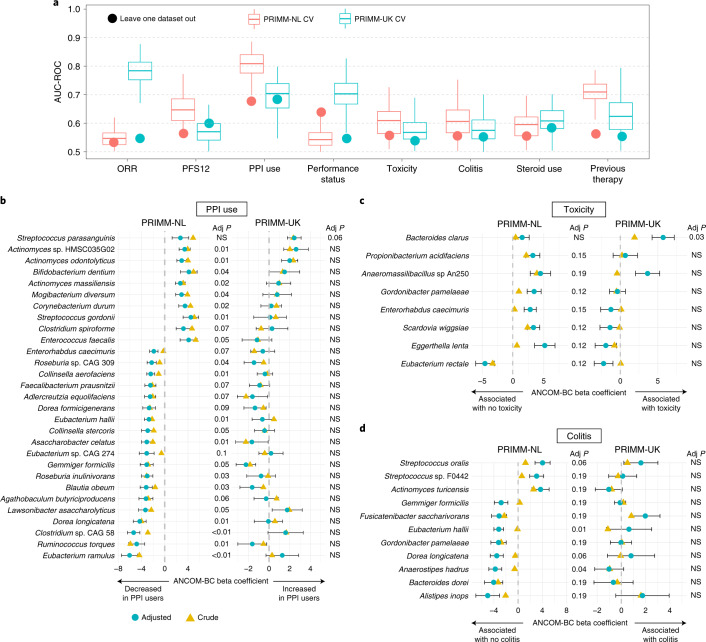
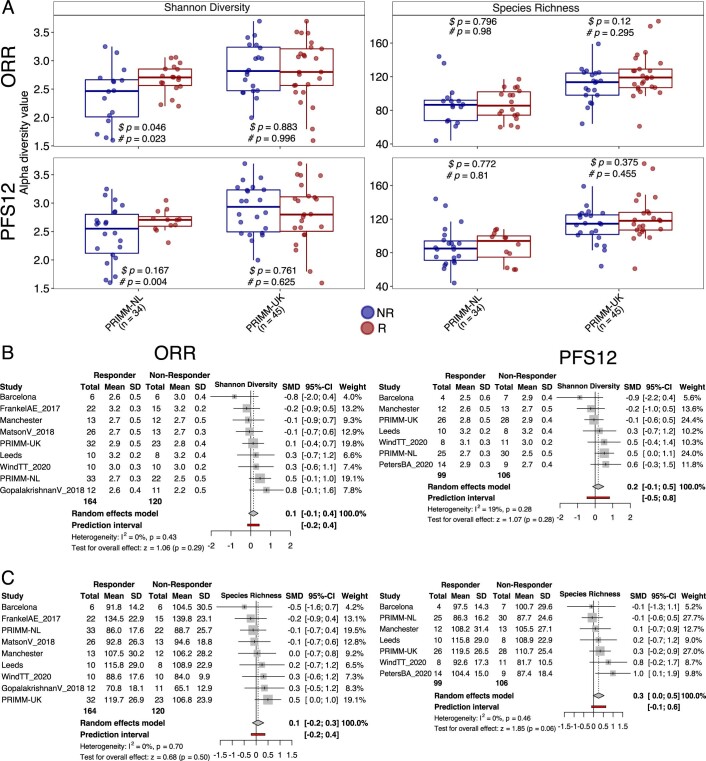
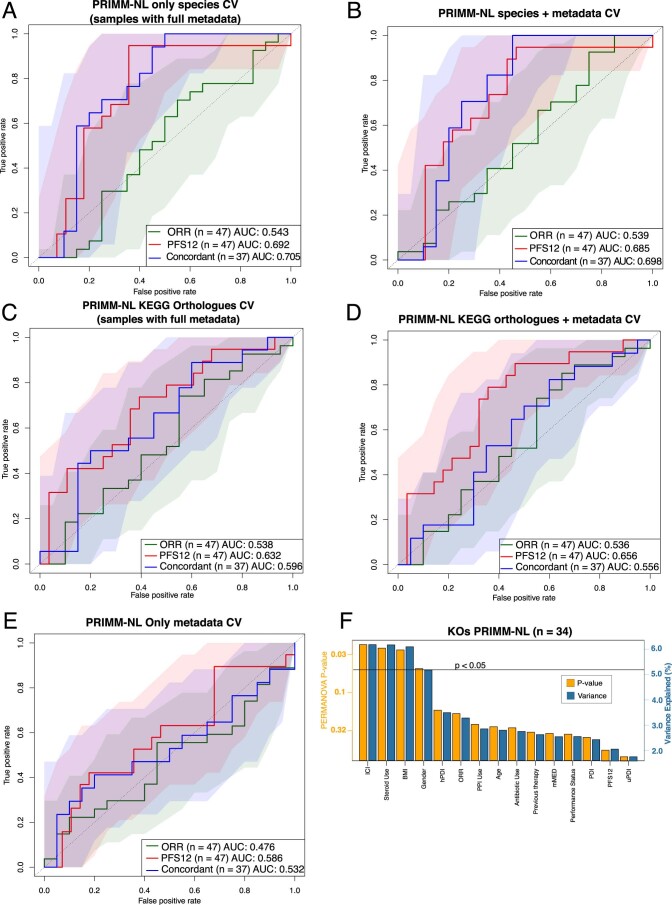
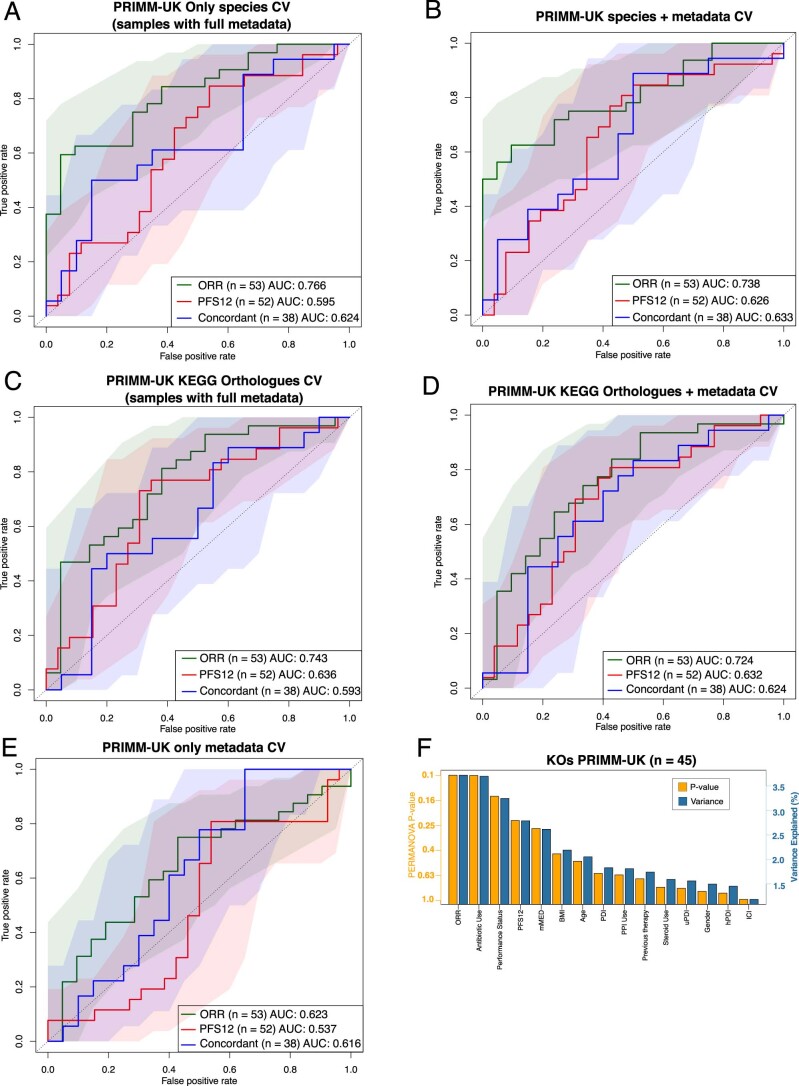
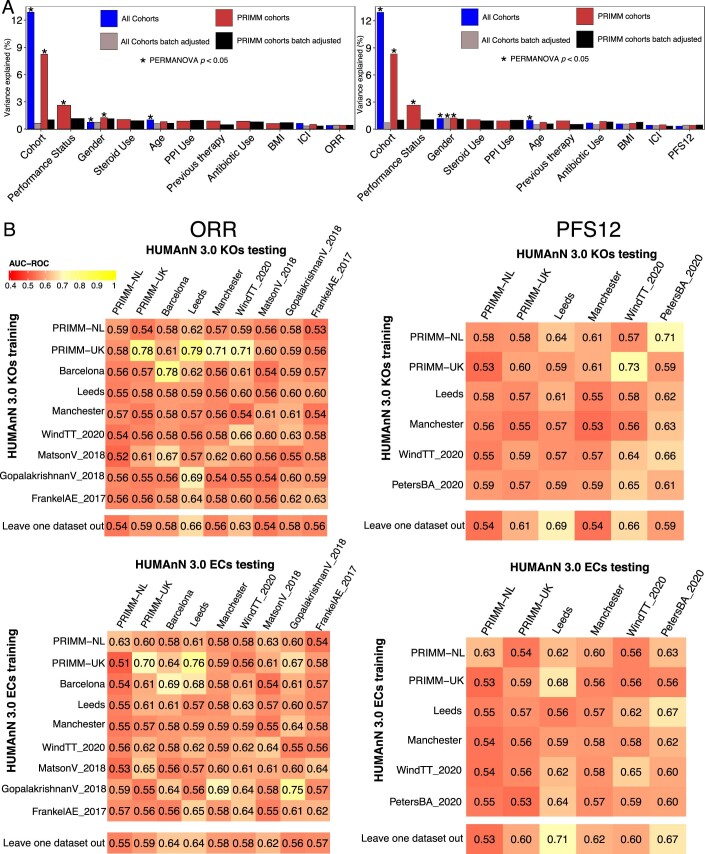
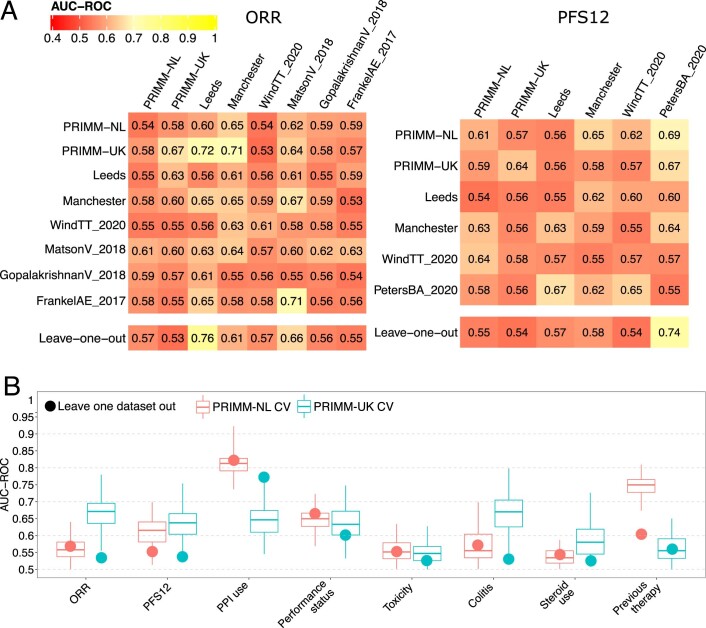
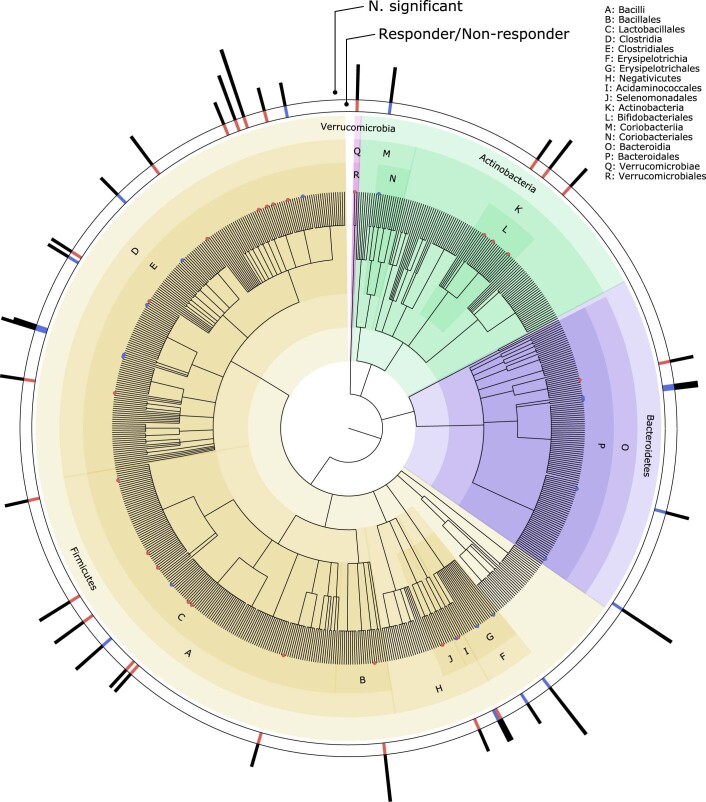
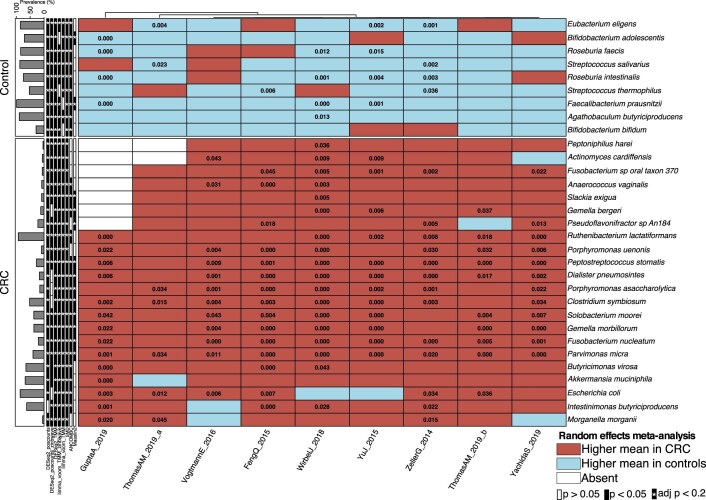
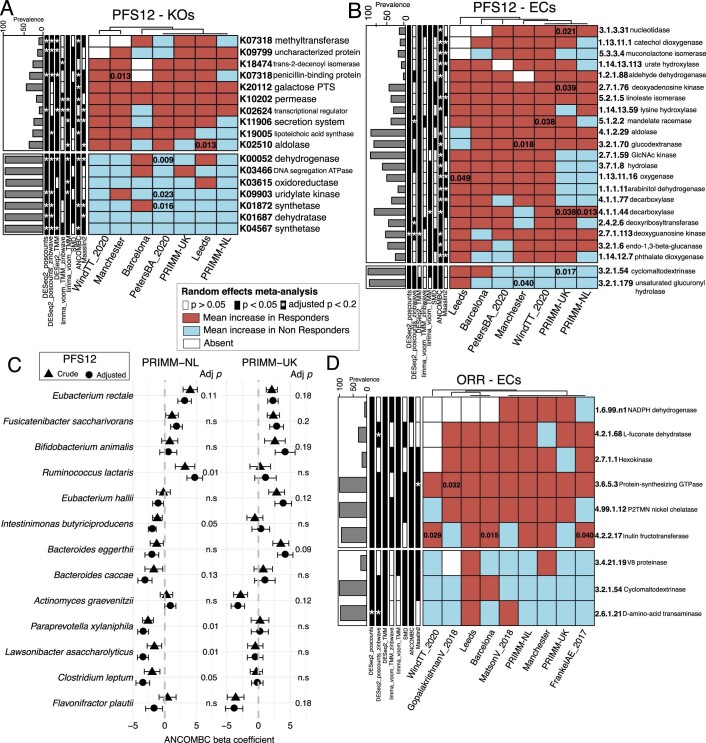
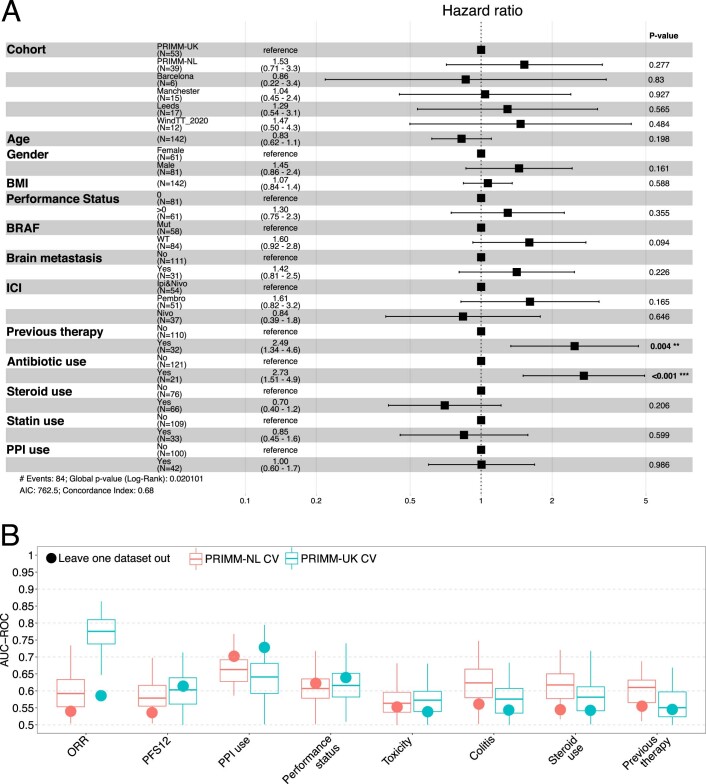
The composition of the gut microbiome has been associated with clinical responses to immune checkpoint inhibitor (ICI) treatment, but there is limited consensus on the specific microbiome characteristics linked to the clinical benefits of ICIs. We performed shotgun metagenomic sequencing of stool samples collected before ICI initiation from five observational cohorts recruiting ICI-naive patients with advanced cutaneous melanoma (n = 165). Integrating the dataset with 147 metagenomic samples from previously published studies, we found that the gut microbiome has a relevant, but cohort-dependent, association with the response to ICIs. A machine learning analysis confirmed the link between the microbiome and overall response rates (ORRs) and progression-free survival (PFS) with ICIs but also revealed limited reproducibility of microbiome-based signatures across cohorts. Accordingly, a panel of species, including Bifidobacterium pseudocatenulatum, Roseburia spp. and Akkermansia muciniphila, associated with responders was identified, but no single species could be regarded as a fully consistent biomarker across studies. Overall, the role of the human gut microbiome in ICI response appears more complex than previously thought, extending beyond differing microbial species simply present or absent in responders and nonresponders. Future studies should adopt larger sample sizes and take into account the complex interplay of clinical factors with the gut microbiome over the treatment course.
© 2022. The Author(s).
Conflict of interest statement
R.K.W. acted as a consultant for Takeda; received unrestricted research grants from Takeda, Johnson & Johnson, Tramedico and Ferring; and received speaker fees from MSD, AbbVie and Janssen Pharmaceuticals. E.R.L. is a consultant for ZOE Global. E.G.E.d.V. reports an advisory role at Daiichi Sankyo, NSABP and Sanofi (paid to University Medical Center Groningen) and research funding from Amgen, AstraZeneca, Bayer, Chugai Pharma, CytomX Therapeutics, G1 Therapeutics, Genentech, Nordic Nanovector, Radius Health, Regeneron, Roche, Servier and Synthon (paid to University Medical Center Groningen). S.P. received speaker fees from Almirall, BMS, ISDIN, La Roche Posay, Leo Pharma, Regeneron, Roche and Sanofi; acted as advisory board member of Almirall, ISDIN, La Roche Posay, Pfizer, Roche, Regeneron, Sanofi and Sun Pharma; and received research funding from Abbie, AMGEN, ISDIN, La Roche Posay, Leo Pharma and Novartis. R.B. has received honoraria from, and sits on advisory boards of, Novartis, BMS and MSD. All other authors declare no competing interests.
Figures
Comment in
-
Gut microbes as biomarkers of ICI response - sharpening the focus.Nat Rev Clin Oncol. 2022 Aug;19(8):495-496. doi: 10.1038/s41571-022-00634-0. Nat Rev Clin Oncol. 2022. PMID: 35449306 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
