

Intestinal microbiota signatures of clinical response and immune-related adverse events in melanoma patients treated with anti-PD-1
- PMID: 35228752
- PMCID: PMC10246505
- DOI: 10.1038/s41591-022-01698-2
Intestinal microbiota signatures of clinical response and immune-related adverse events in melanoma patients treated with anti-PD-1
Abstract
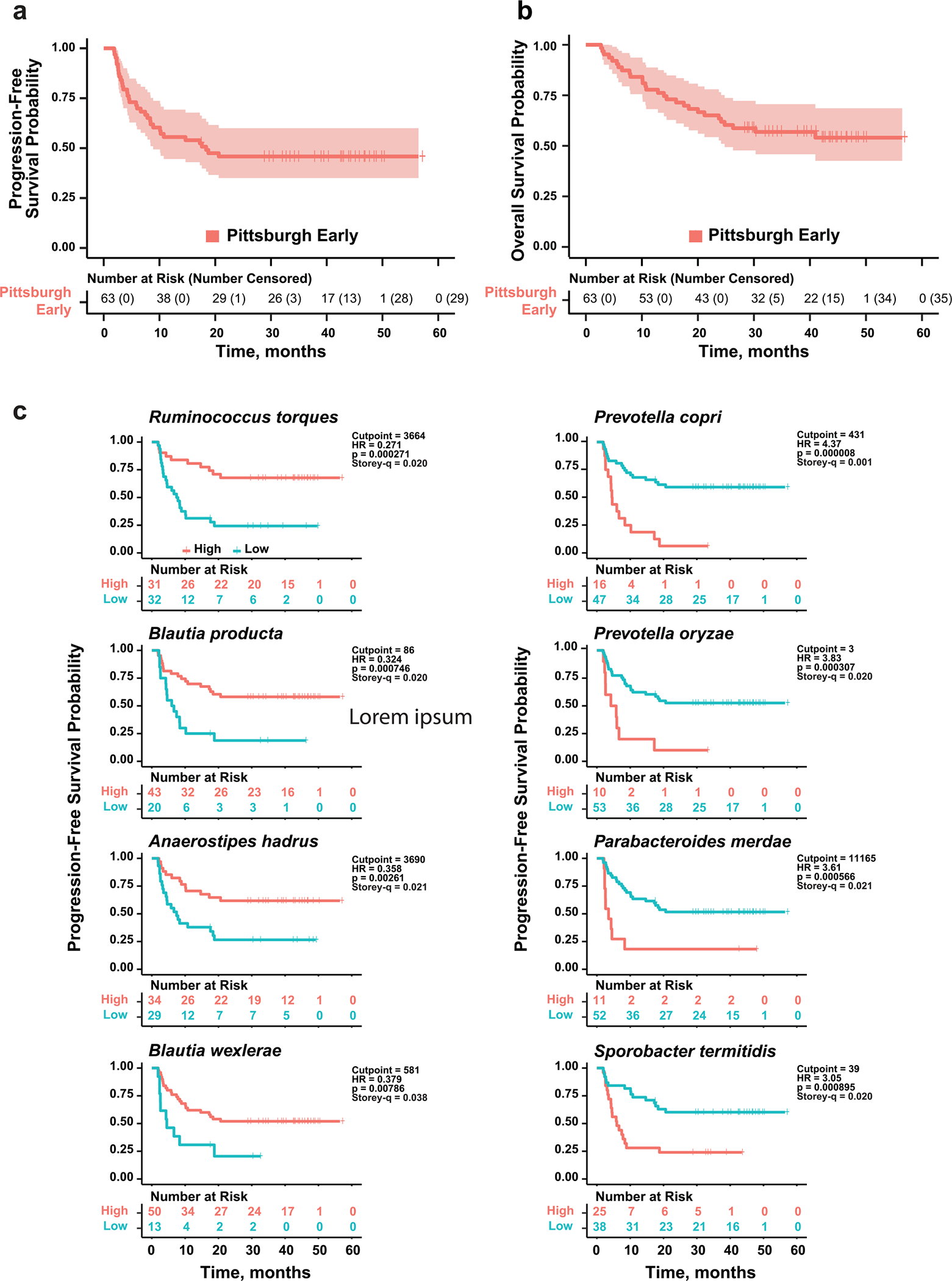
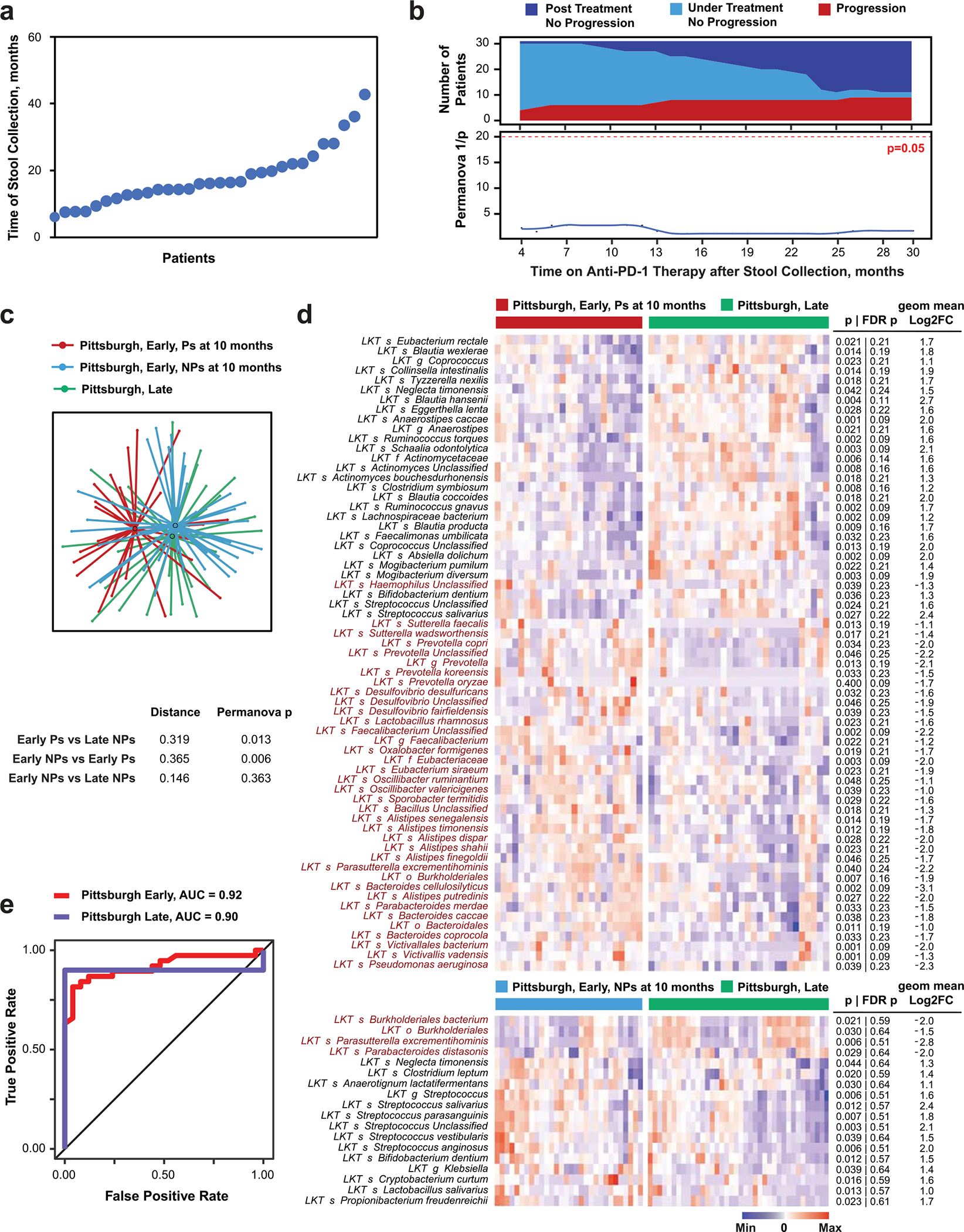
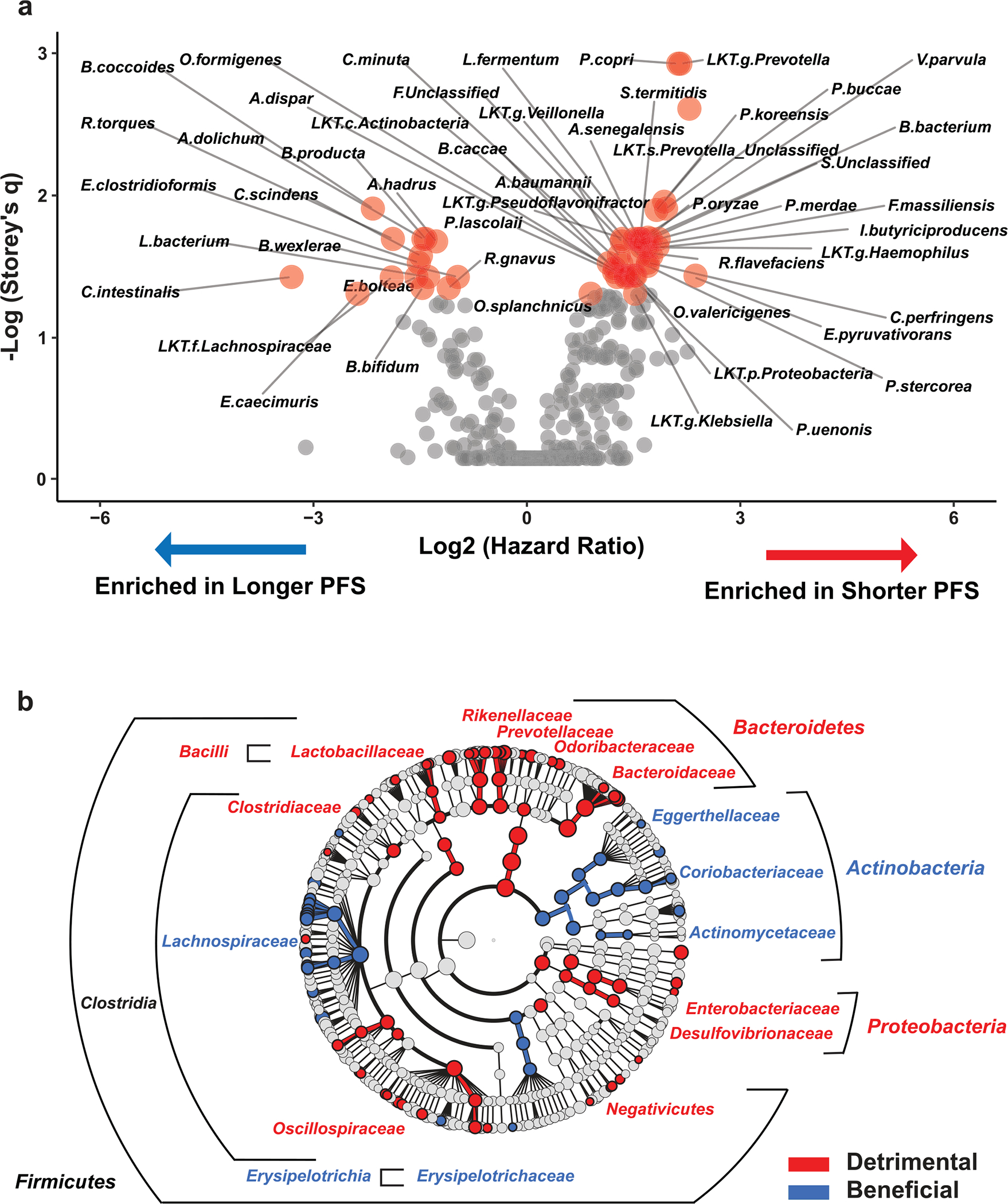
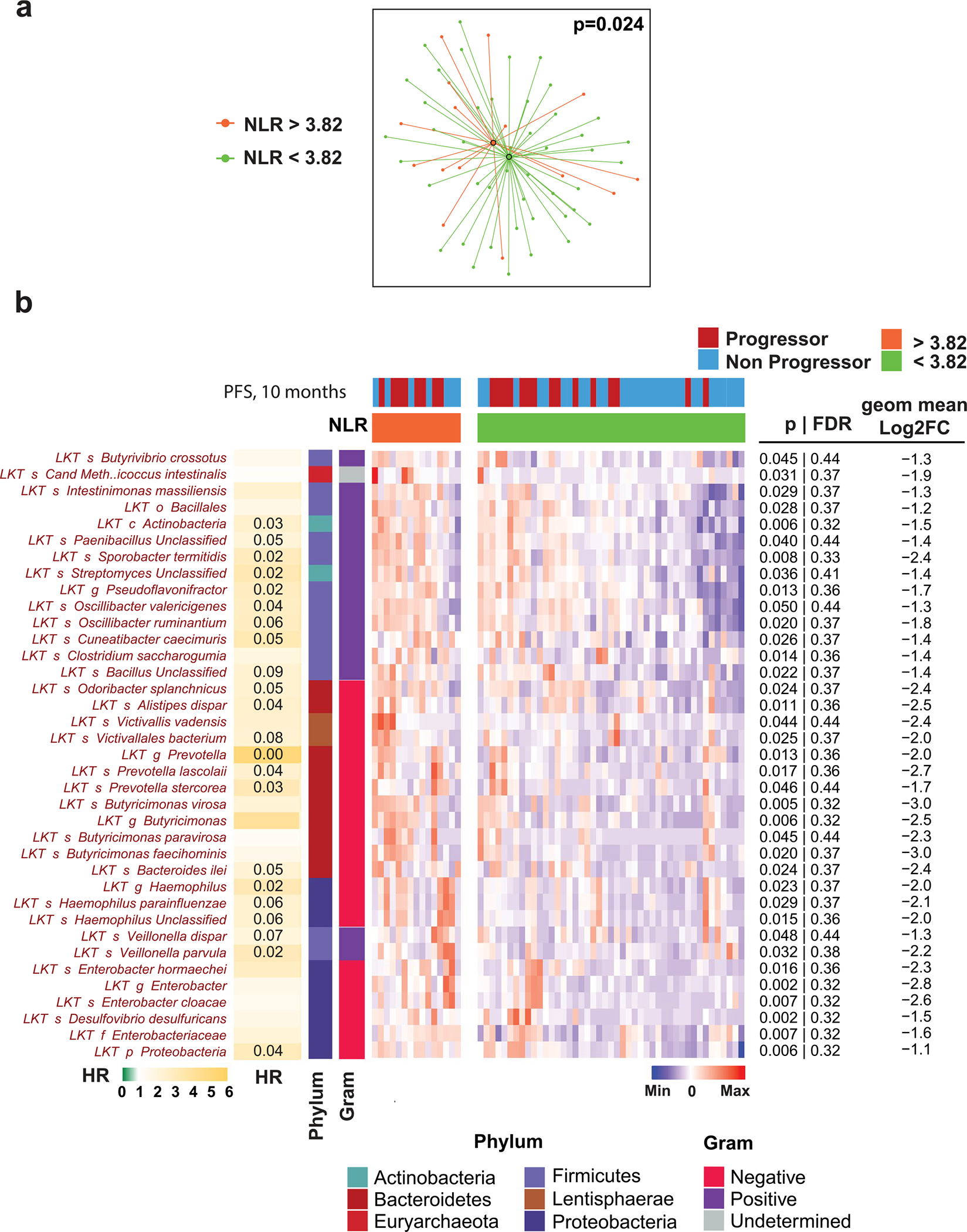
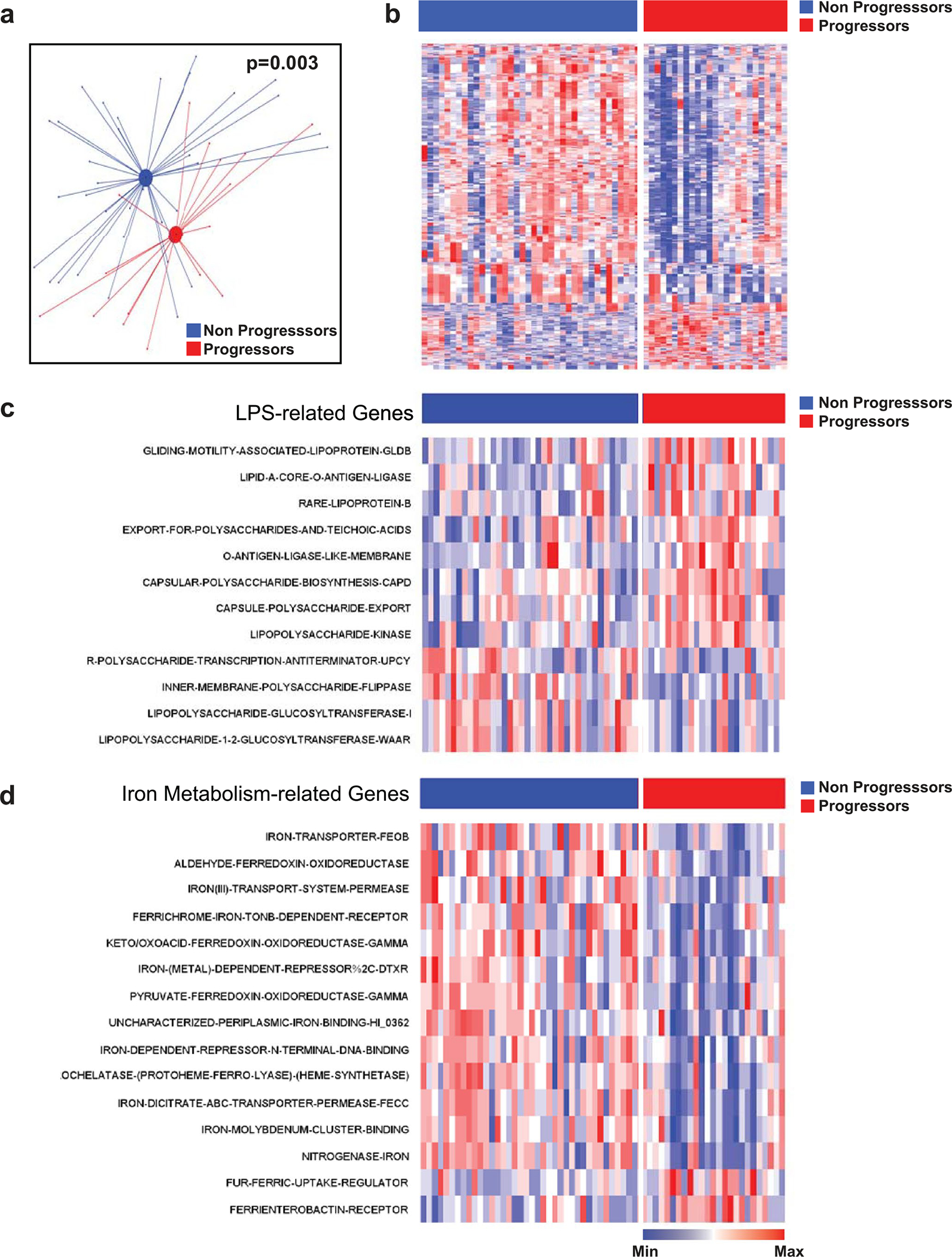
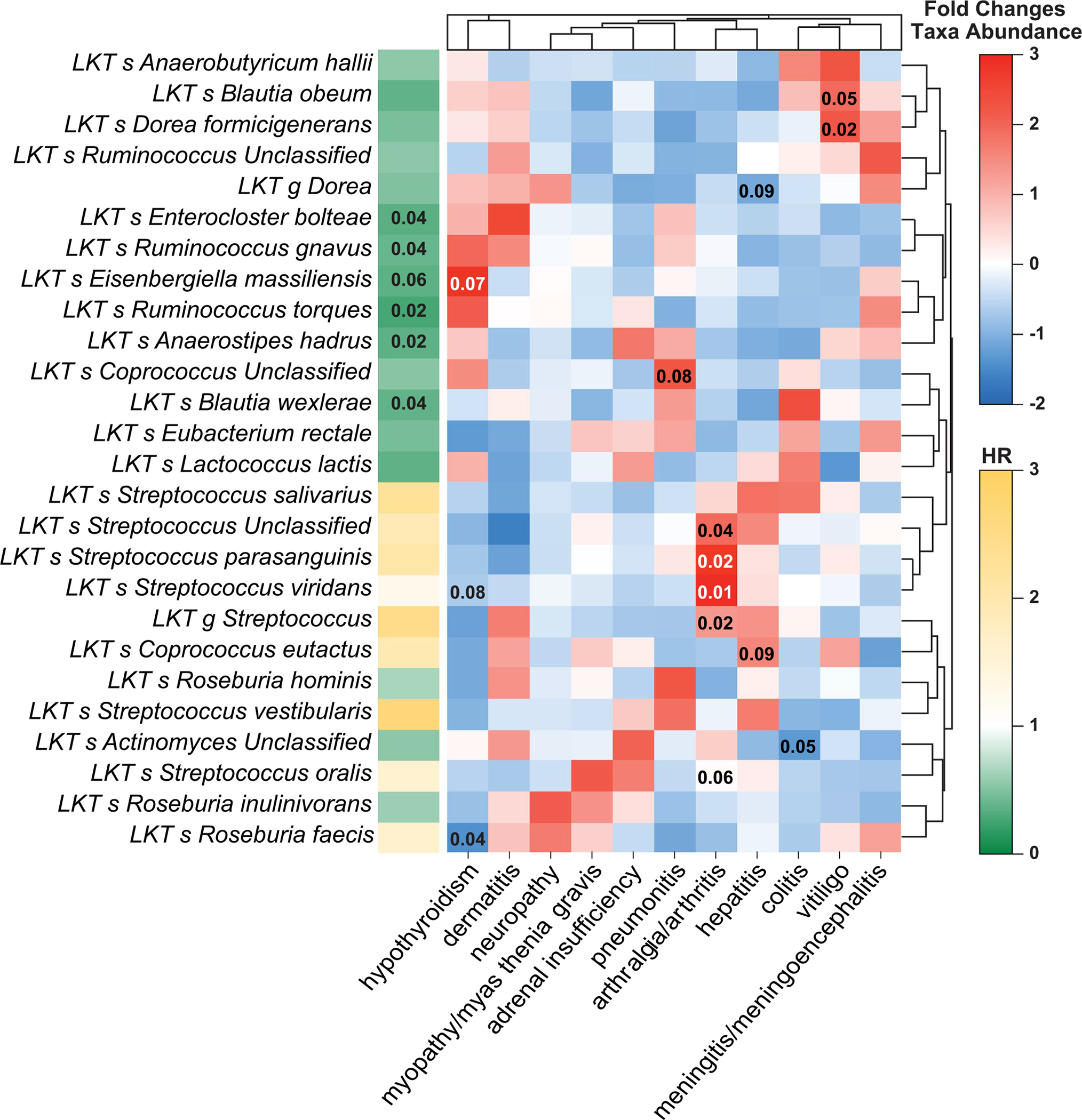
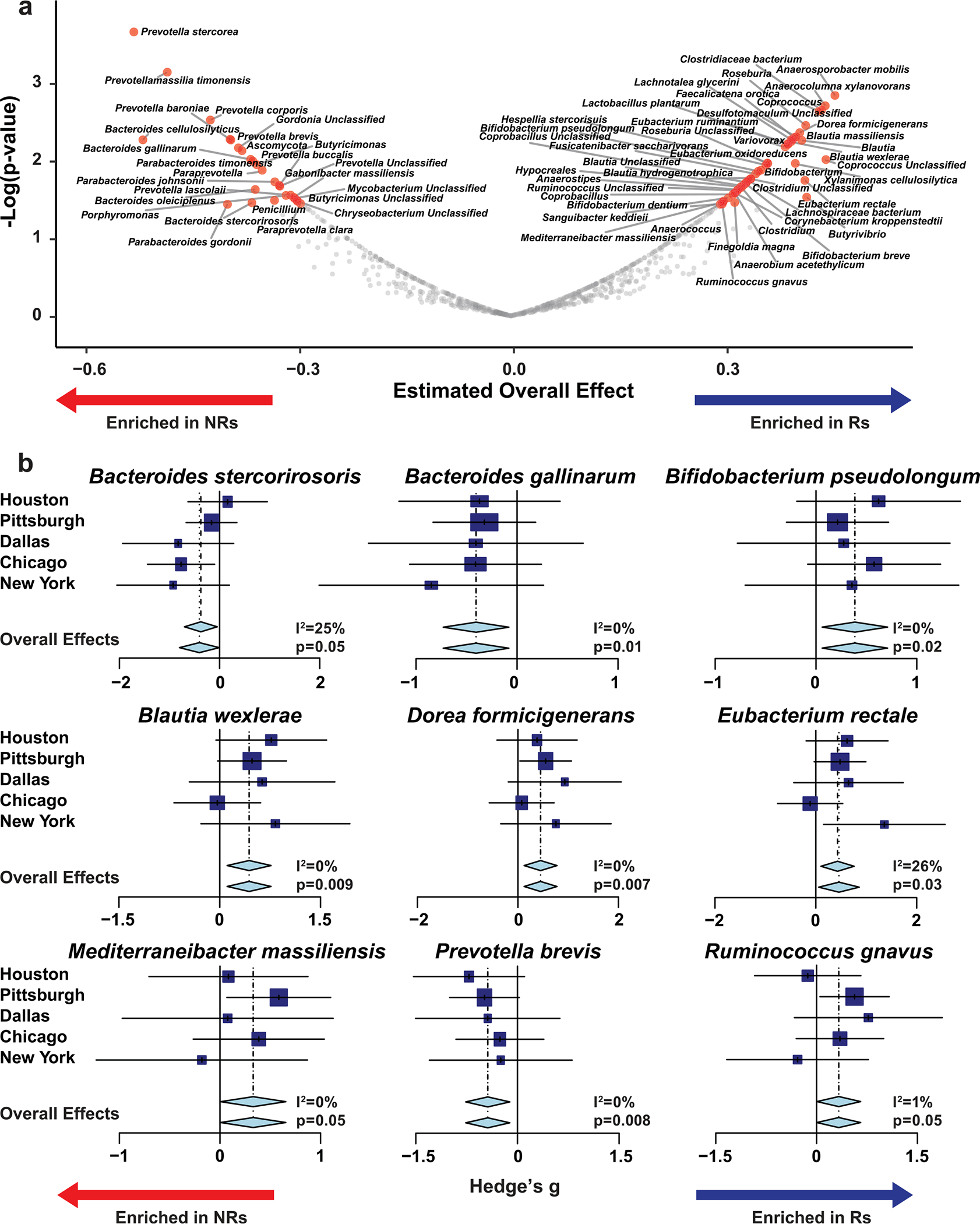
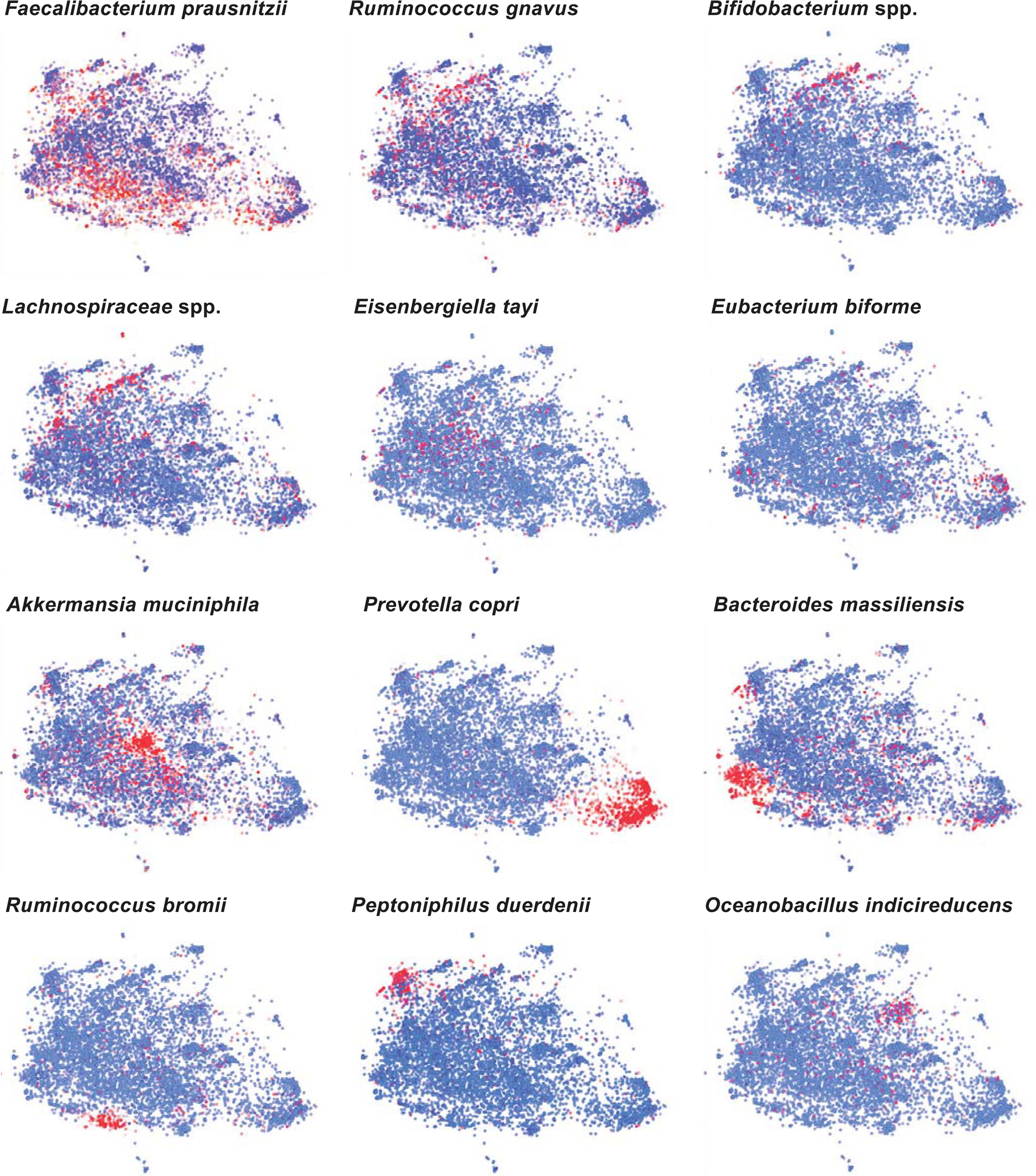
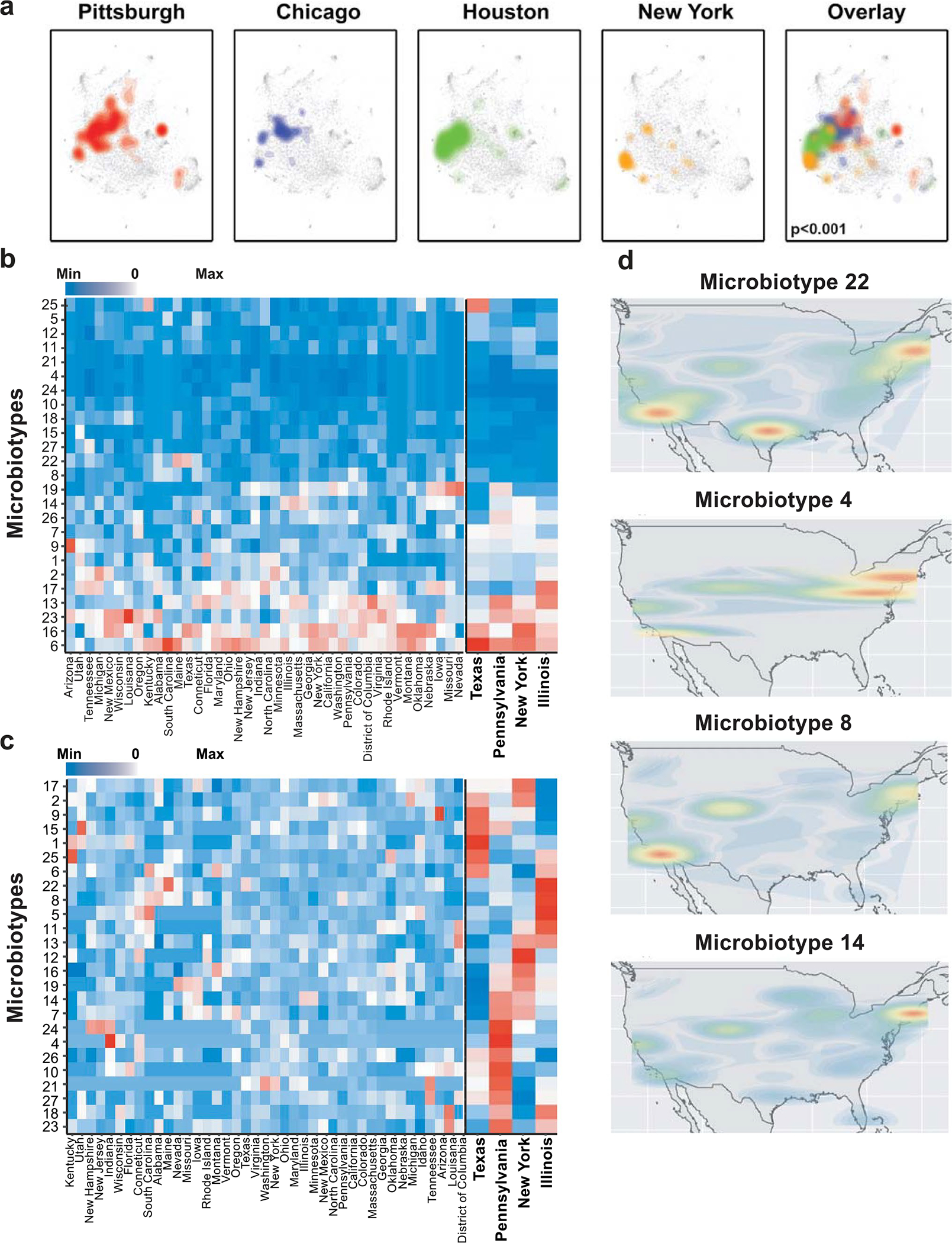
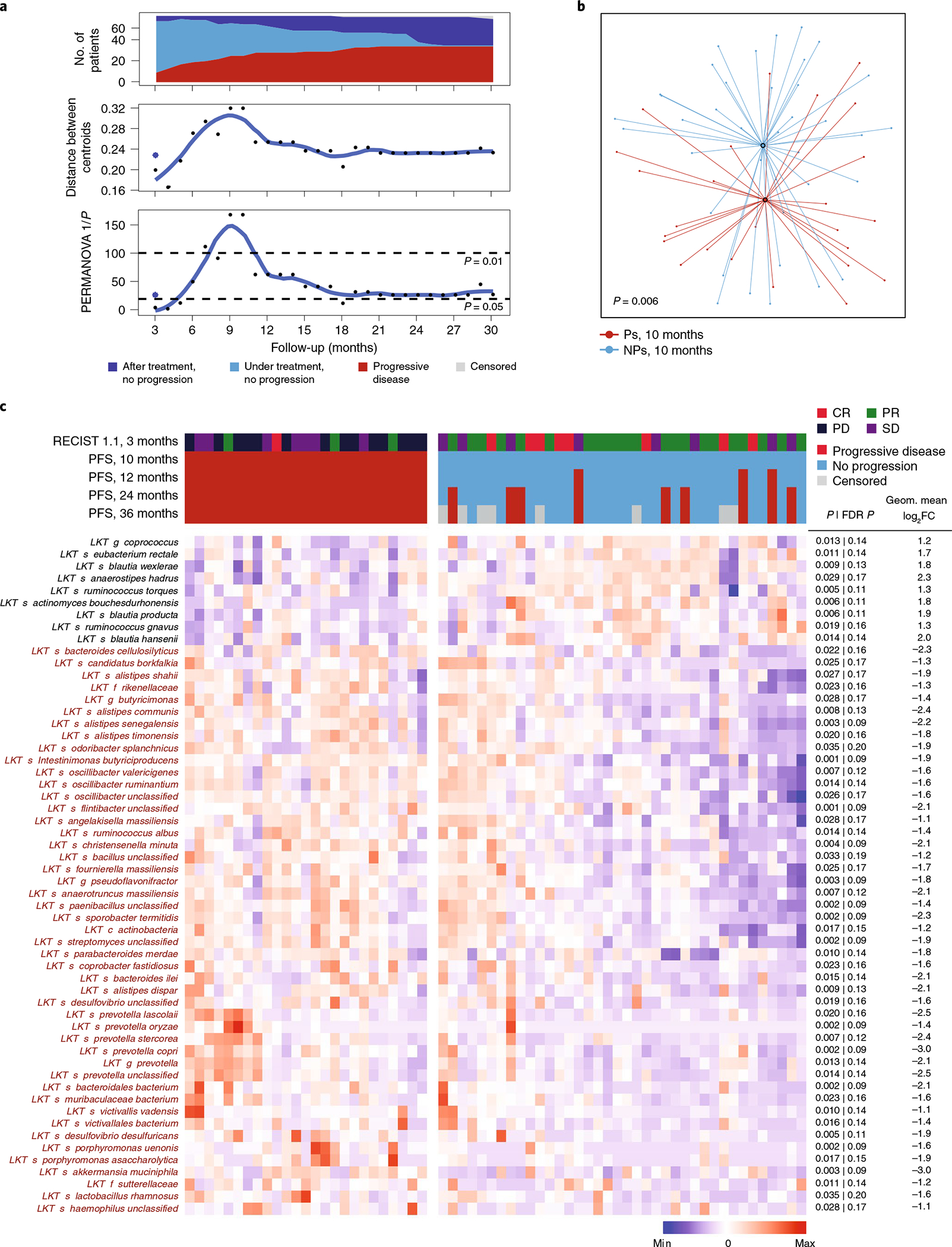
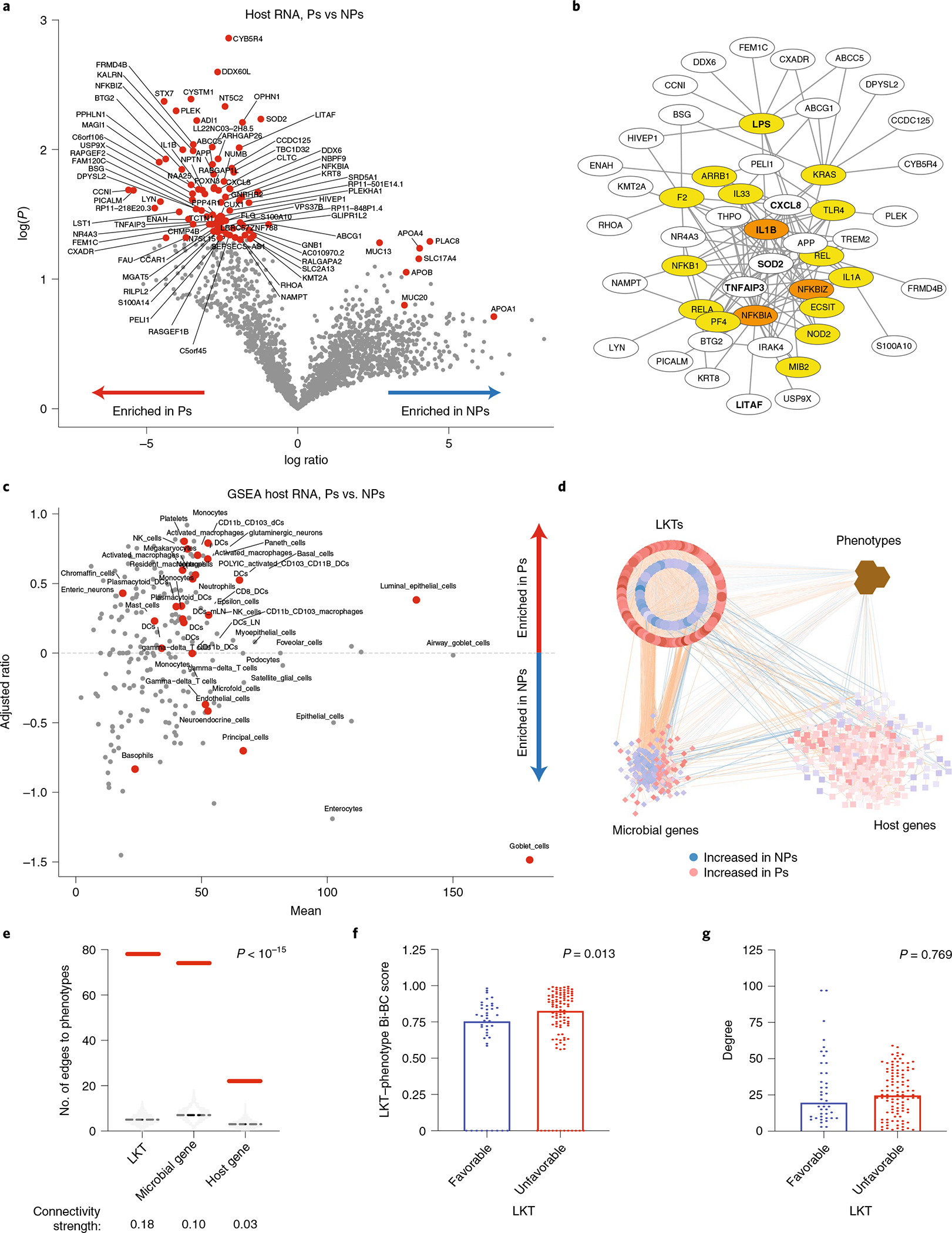
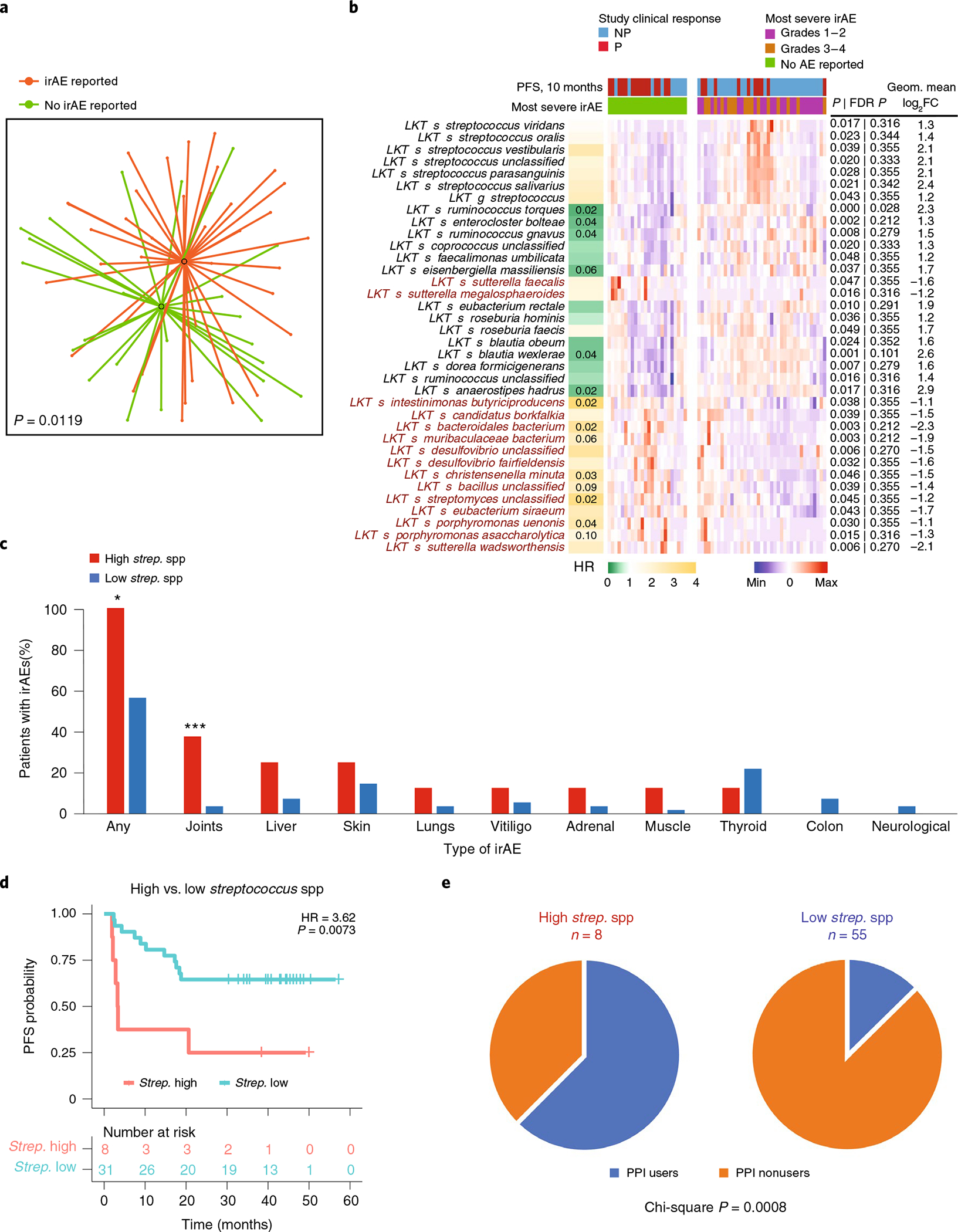
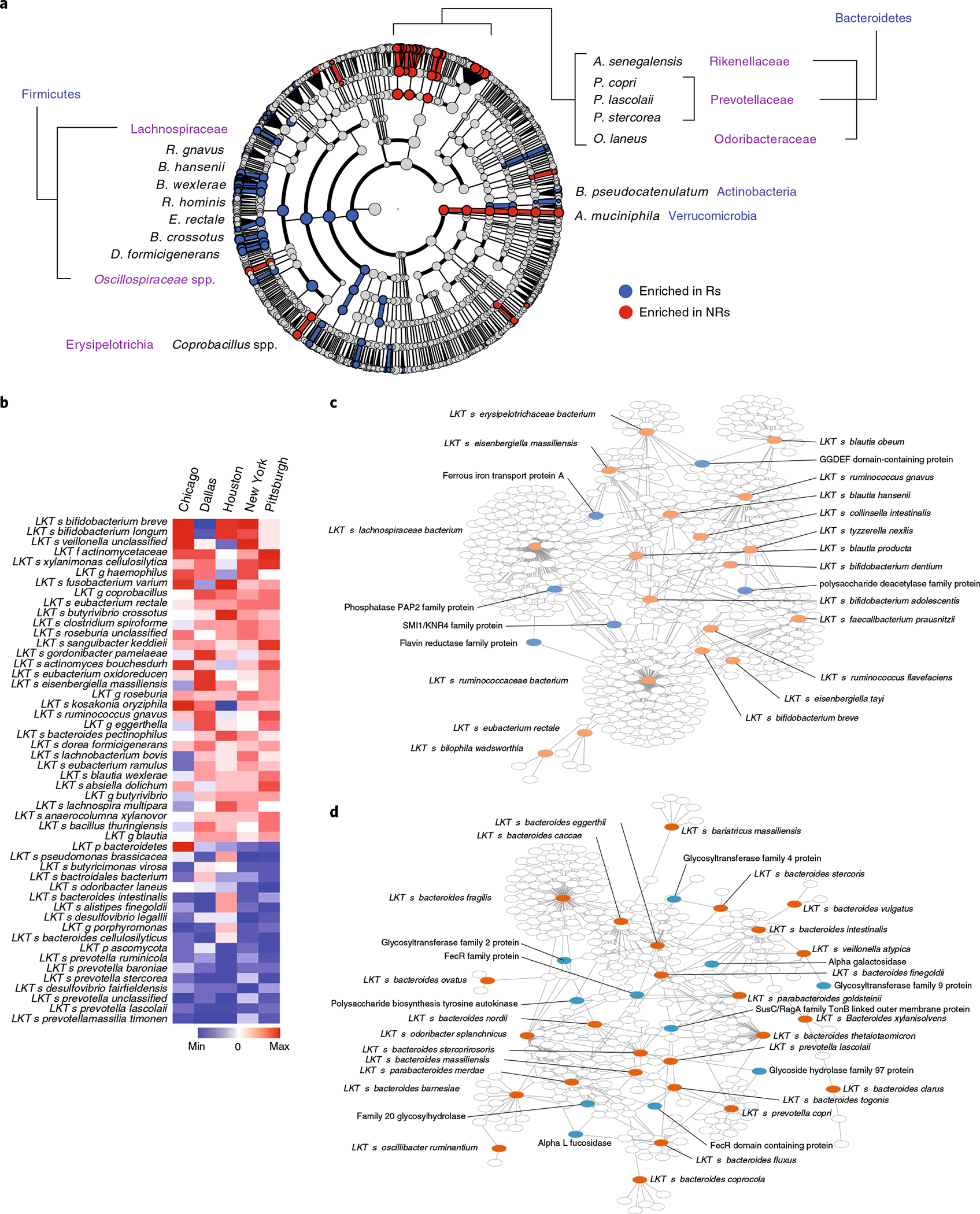
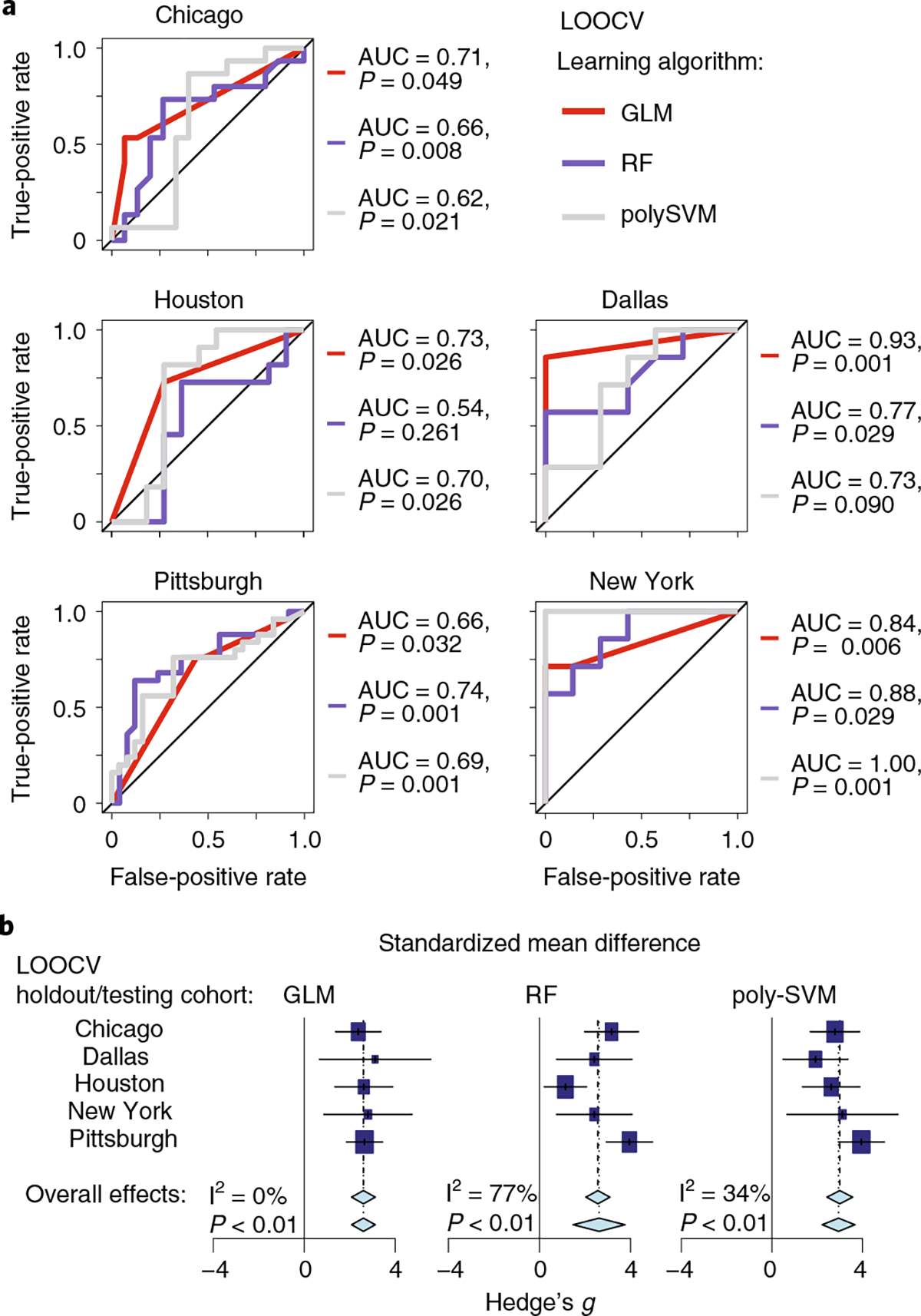
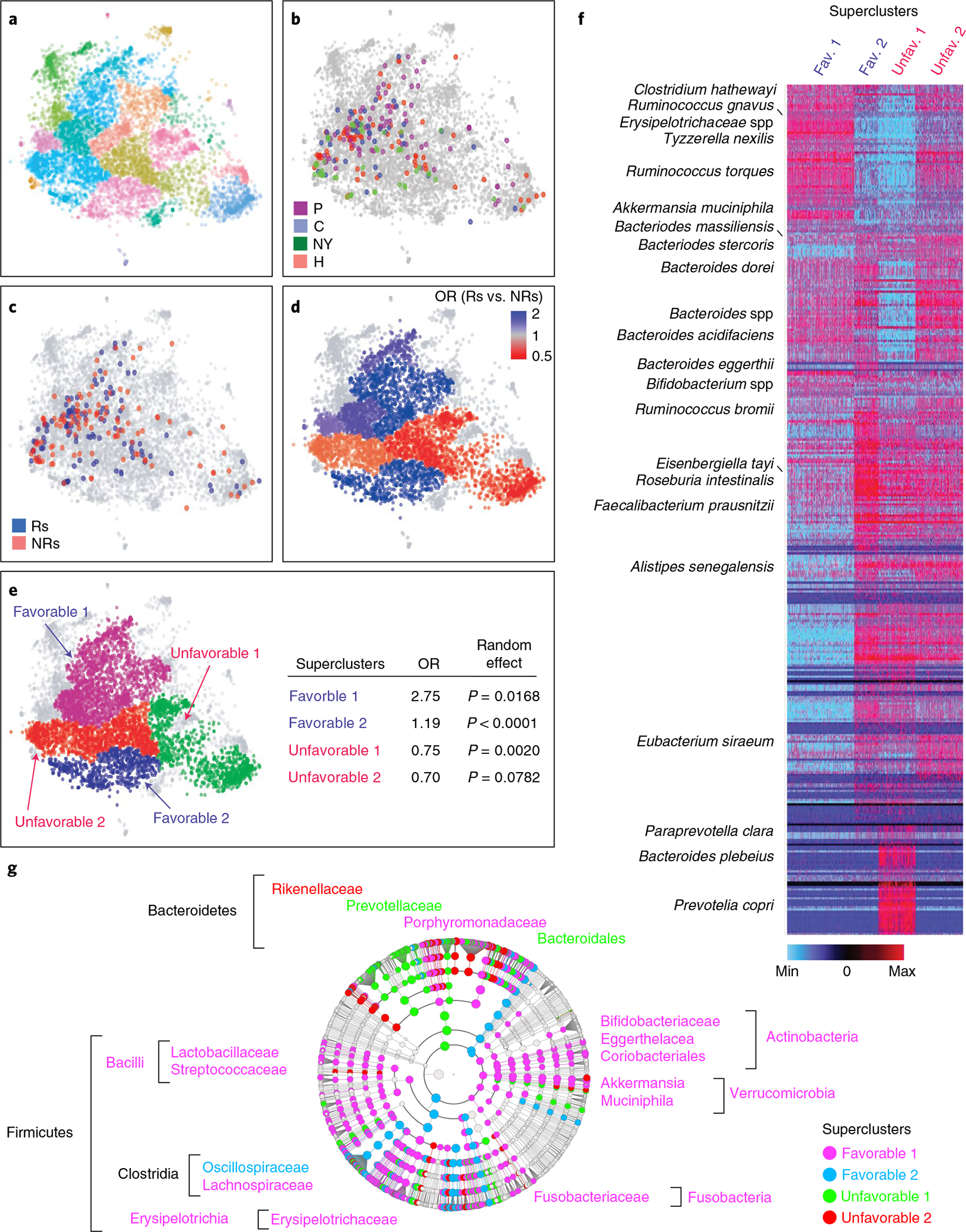
Ample evidence indicates that the gut microbiome is a tumor-extrinsic factor associated with antitumor response to anti-programmed cell death protein-1 (PD-1) therapy, but inconsistencies exist between published microbial signatures associated with clinical outcomes. To resolve this, we evaluated a new melanoma cohort, along with four published datasets. Time-to-event analysis showed that baseline microbiota composition was optimally associated with clinical outcome at approximately 1 year after initiation of treatment. Meta-analysis and other bioinformatic analyses of the combined data show that bacteria associated with favorable response are confined within the Actinobacteria phylum and the Lachnospiraceae/Ruminococcaceae families of Firmicutes. Conversely, Gram-negative bacteria were associated with an inflammatory host intestinal gene signature, increased blood neutrophil-to-lymphocyte ratio, and unfavorable outcome. Two microbial signatures, enriched for Lachnospiraceae spp. and Streptococcaceae spp., were associated with favorable and unfavorable clinical response, respectively, and with distinct immune-related adverse effects. Despite between-cohort heterogeneity, optimized all-minus-one supervised learning algorithms trained on batch-corrected microbiome data consistently predicted outcomes to programmed cell death protein-1 therapy in all cohorts. Gut microbial communities (microbiotypes) with nonuniform geographical distribution were associated with favorable and unfavorable outcomes, contributing to discrepancies between cohorts. Our findings shed new light on the complex interaction between the gut microbiome and response to cancer immunotherapy, providing a roadmap for future studies.
© 2022. This is a U.S. government work and not under copyright protection in the U.S.; foreign copyright protection may apply.
Conflict of interest statement
Competing interests
D.D. reports the following disclosures: Arcus, Bristol-Myers Squibb, Checkmate Pharmaceuticals, CellSight Technologies, Merck, GlaxoSmithKline/Tesaro (research support); Array Biopharma, Checkmate Pharmaceuticals, Finch, Incyte, Immunocore, Merck; Shionogi (consulting); and Vedanta Biosciences (scientific advisory board). H.M.Z. reports the following disclosures: Bristol-Myers Squibb, Checkmate Pharmaceuticals, GlaxoSmithKline (research support); Bristol-Myers Squibb, Checkmate Pharmaceuticals, GlaxoSmithKline, Vedanta (consulting). D.D., H.M.Z., J.A.M., R.R.R., G.T. and A.K.D. are inventors on a patent application (US patent no. 63/208,719) submitted by the University of Pittsburgh that covers methods to enhance checkpoint blockade therapy by the microbiome. The other authors declare no competing interests.
Figures

Comment in
-
Gut microbes as biomarkers of ICI response - sharpening the focus.Nat Rev Clin Oncol. 2022 Aug;19(8):495-496. doi: 10.1038/s41571-022-00634-0. Nat Rev Clin Oncol. 2022. PMID: 35449306 No abstract available.
References
-
- Dzutsev A, Goldszmid RS, Viaud S, Zitvogel L & Trinchieri G The role of the microbiota in inflammation, carcinogenesis, and cancer therapy. Eur. J. Immunol. 45, 17–31 (2015). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
