

Deep-sequence phylogenetics to quantify patterns of HIV transmission in the context of a universal testing and treatment trial - BCPP/Ya Tsie trial
- PMID: 35229714
- PMCID: PMC8912920
- DOI: 10.7554/eLife.72657
Deep-sequence phylogenetics to quantify patterns of HIV transmission in the context of a universal testing and treatment trial - BCPP/Ya Tsie trial
Abstract
Background: Mathematical models predict that community-wide access to HIV testing-and-treatment can rapidly and substantially reduce new HIV infections. Yet several large universal test-and-treat HIV prevention trials in high-prevalence epidemics demonstrated variable reduction in population-level incidence.
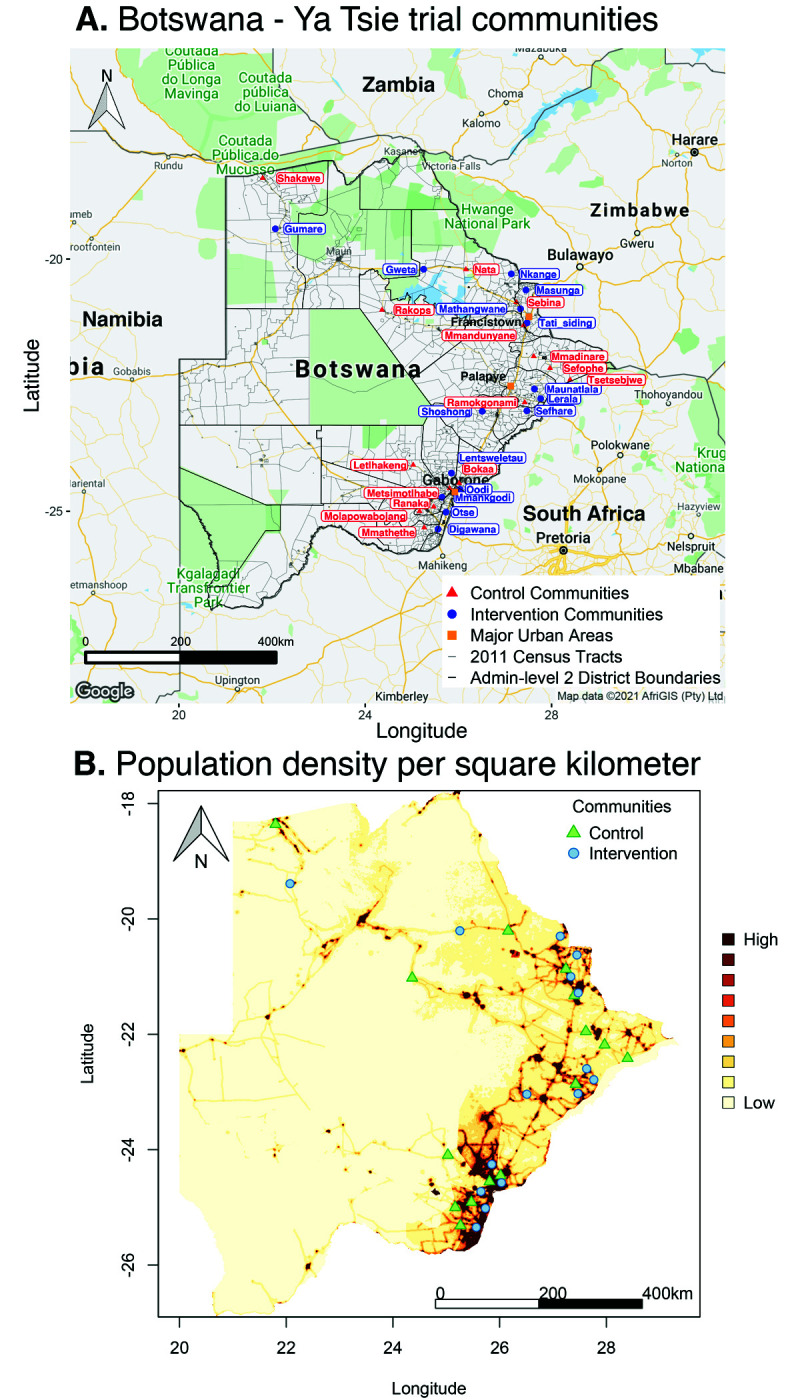
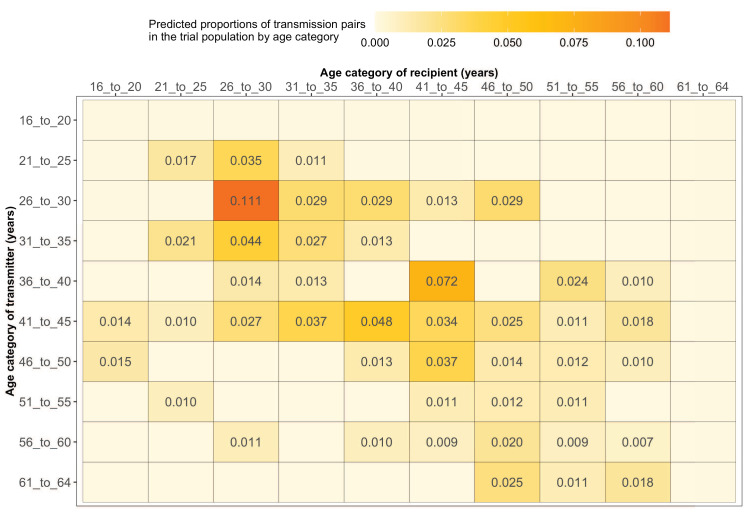
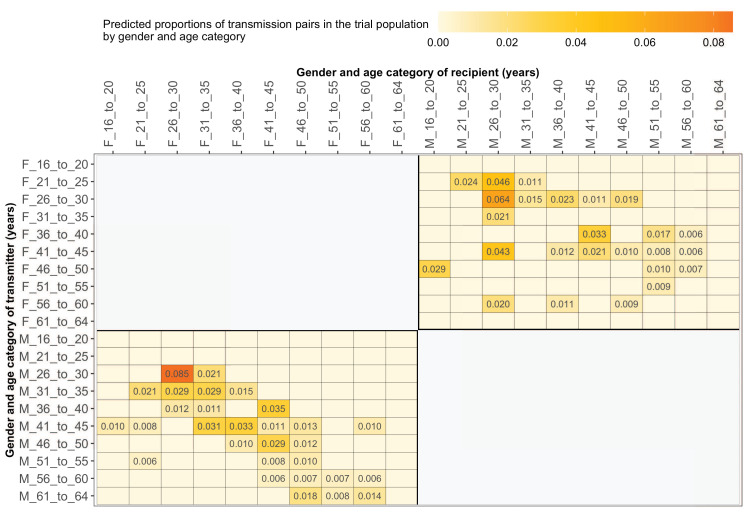
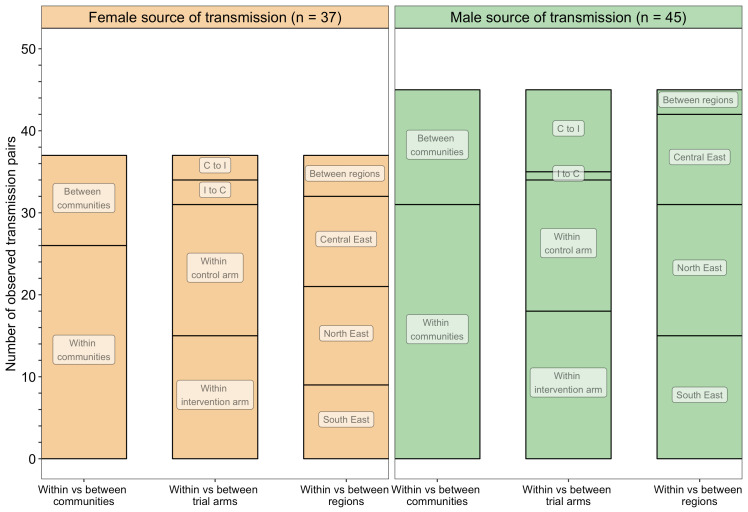
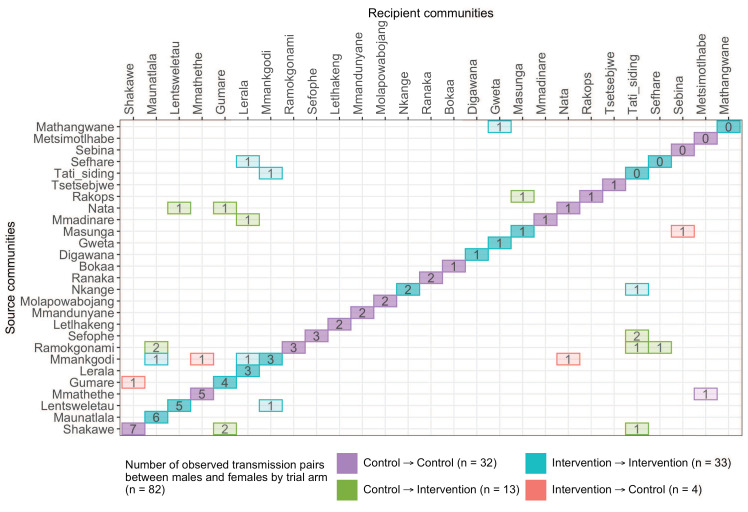
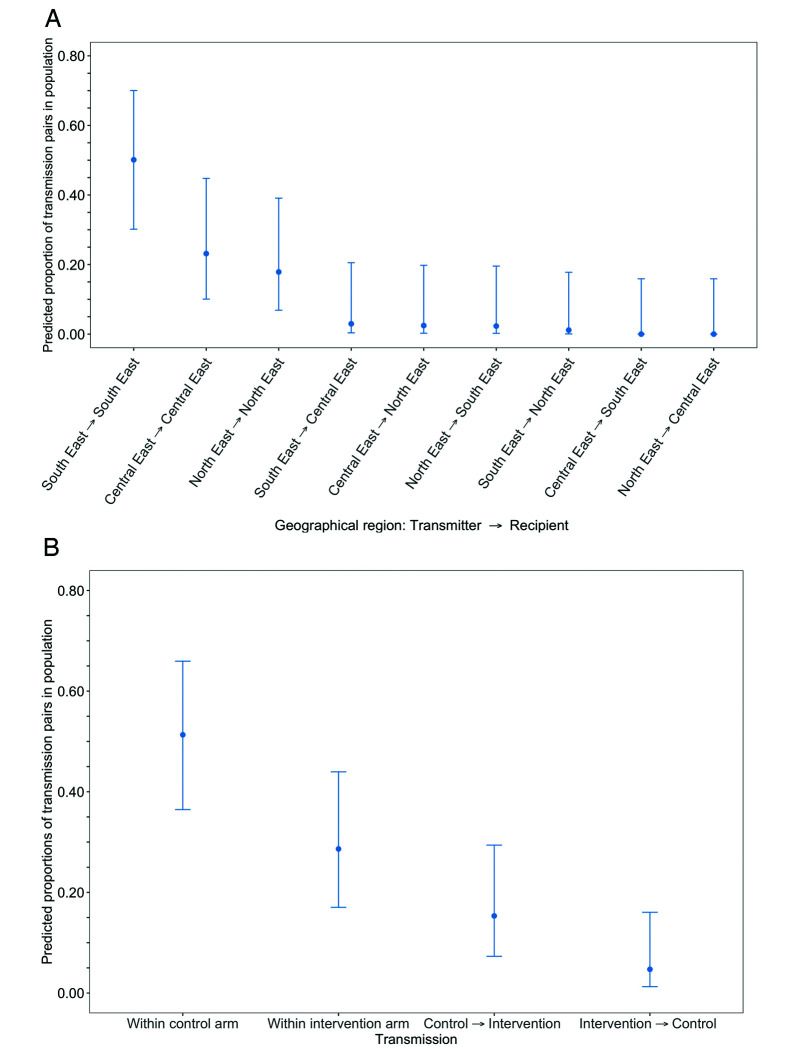
Methods: To elucidate patterns of HIV spread in universal test-and-treat trials, we quantified the contribution of geographic-location, gender, age, and randomized-HIV-intervention to HIV transmissions in the 30-community Ya Tsie trial in Botswana. We sequenced HIV viral whole genomes from 5114 trial participants among the 30 trial communities.
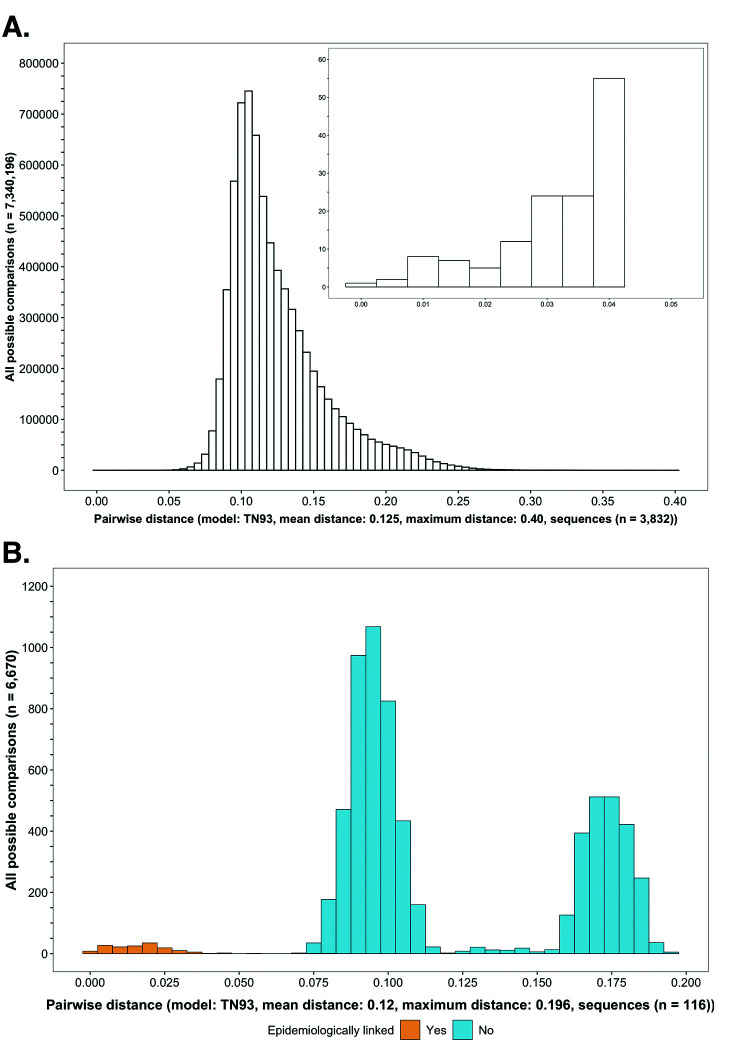
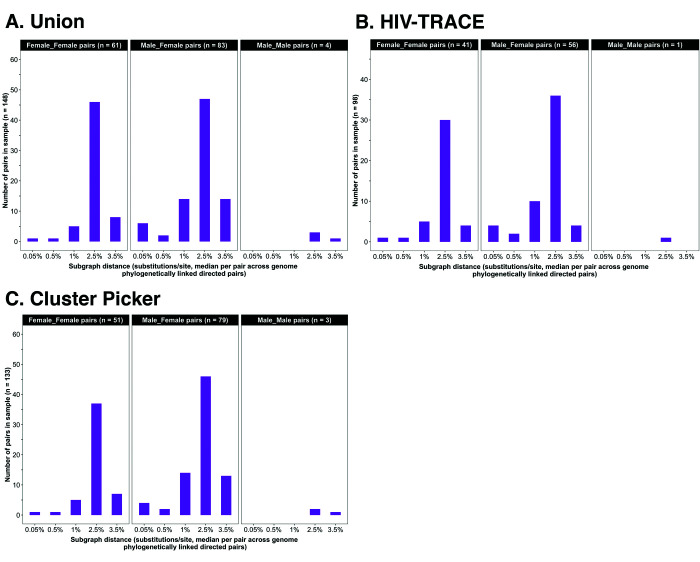
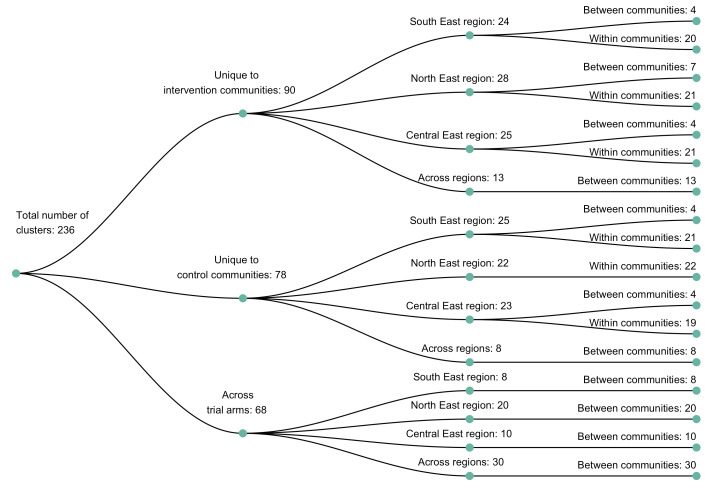
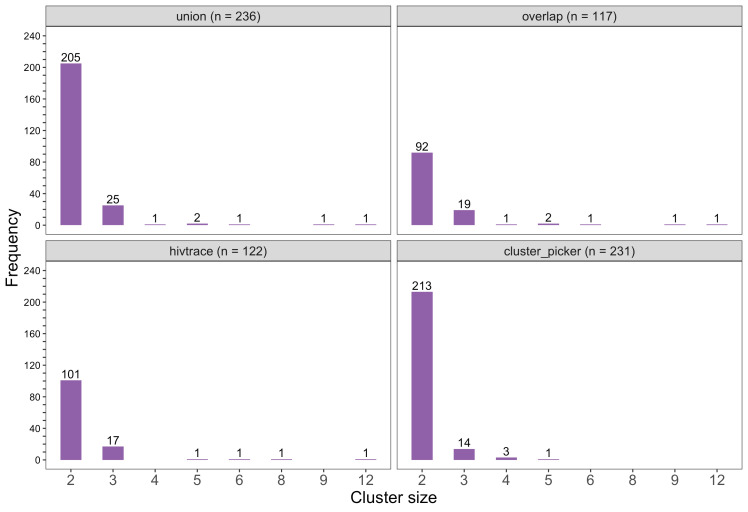
Results: Deep-sequence phylogenetic analysis revealed that most inferred HIV transmissions within the trial occurred within the same or between neighboring communities, and between similarly aged partners. Transmissions into intervention communities from control communities were more common than the reverse post-baseline (30% [12.2 - 56.7] vs. 3% [0.1 - 27.3]) than at baseline (7% [1.5 - 25.3] vs. 5% [0.9 - 22.9]) compatible with a benefit from treatment-as-prevention.
Conclusions: Our findings suggest that population mobility patterns are fundamental to HIV transmission dynamics and to the impact of HIV control strategies.
Funding: This study was supported by the National Institute of General Medical Sciences (U54GM088558), the Fogarty International Center (FIC) of the U.S. National Institutes of Health (D43 TW009610), and the President's Emergency Plan for AIDS Relief through the Centers for Disease Control and Prevention (CDC) (Cooperative agreements U01 GH000447 and U2G GH001911).
Keywords: HIV prevention; HIV transmission; bumblebee; epidemiology; genetics; genomics; infectious disease; microbiology; phylogenetics; universal test; universal treat; viruses.
Conflict of interest statement
LM, YZ, TG, VD, ET, VN, JM, PB, TS, RL, MP, SM, JM, CF, ME No competing interests declared, SL participates in a data safety monitoring board for NIH-funded study of PK of TB drugs and antiretrovirals in children and on a scientific advisory board for observational study of DTG programmatic rollout in Botswana. Is also a member of the Finance Board and a member of the Board of Directors for the Botswana Harvard AIDS Institute Partnership. Receives no financial compensation for these roles, and has no other competing interests to declare, ML is a Reviewing Editor for eLife. Has received consultancy fees from Merck, University of Virginia Miller Center and Janssen, and has performed unpaid consultancy work for Janssen, Pfizer and Astra Zeneca. Has also received payments or honoraria from Sanofi Pasteur and Bristol Myers Squibb. ML has no other competing interests to declare
Figures
References
-
- Balkus JE, Nair G, Montgomery ET, Mishra A, Palanee-Phillips T, Ramjee G, Panchia R, Selepe P, Richardson BA, Chirenje ZM, Marrazzo JM. Age-Disparate Partnerships and Risk of HIV-1 Acquisition Among South African Women Participating in the VOICE Trial. Journal of Acquired Immune Deficiency Syndromes. 2015;70:212–217. doi: 10.1097/QAI.0000000000000715. - DOI - PMC - PubMed
-
- Chan SK, Thornton LR, Chronister KJ, Meyer J, Wolverton M, Johnson CK, Arafat RR, Joyce PM, Switzer WM, Heneine W, Shankar A, Granade T, Owen MS, Sprinkle P, Sullivan V, Centers for Disease Control and Prevention (CDC) Likely female-to-female sexual transmission of HIV--Texas, 2012. MMWR. Morbidity and Mortality Weekly Report. 2014;63:209–212. - PMC - PubMed
-
- Cherry S. A Comparison of Confidence Interval Methods for Habitat Use-Availability Studies. The Journal of Wildlife Management. 1996;60:653. doi: 10.2307/3802084. - DOI
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
