

Robust and annotation-free analysis of alternative splicing across diverse cell types in mice
- PMID: 35229721
- PMCID: PMC8975553
- DOI: 10.7554/eLife.73520
Robust and annotation-free analysis of alternative splicing across diverse cell types in mice
Abstract
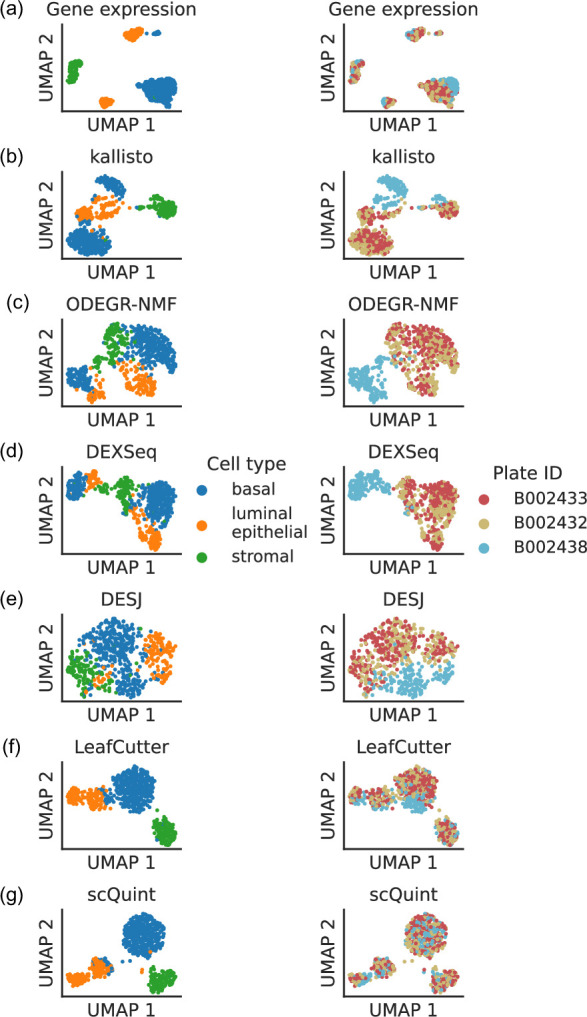
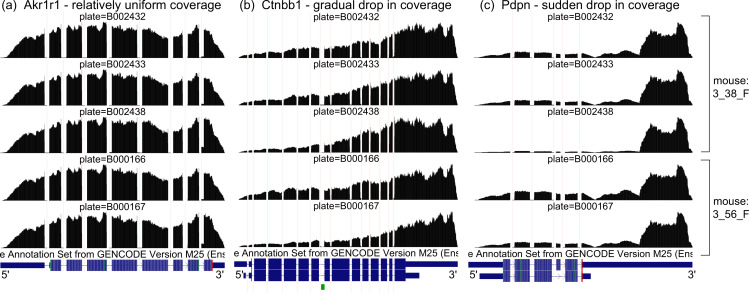
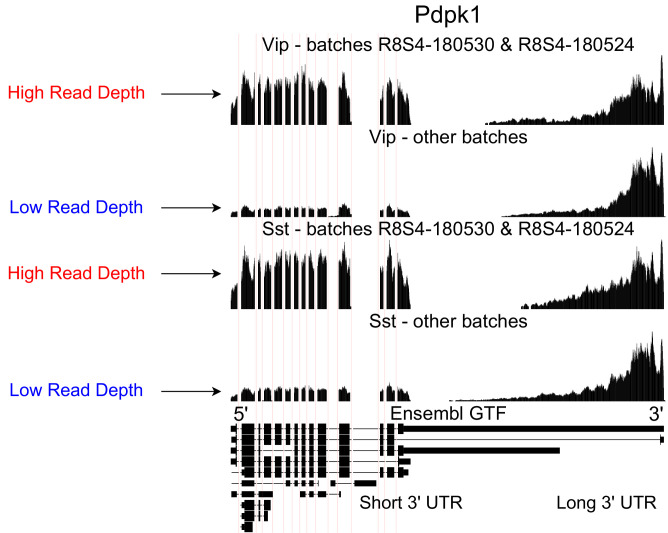
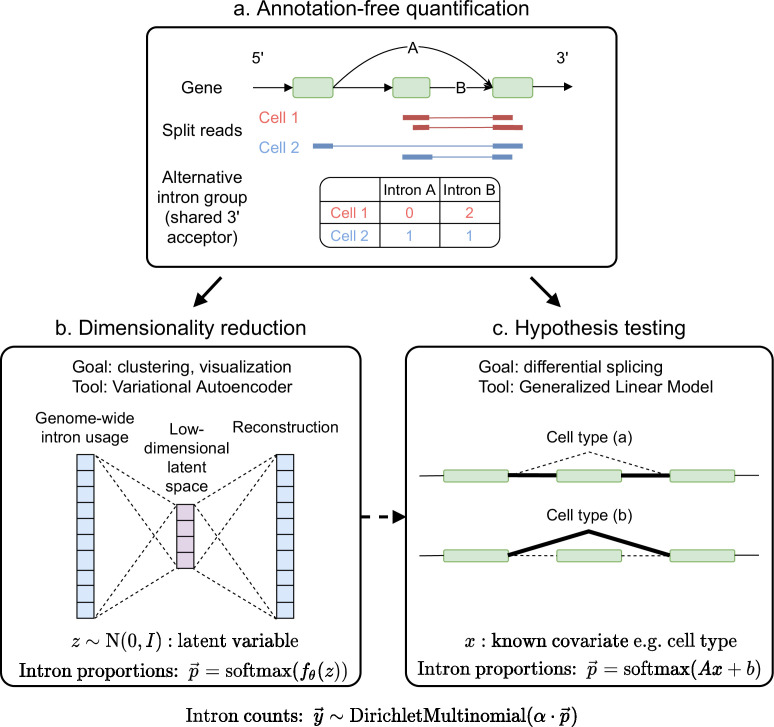
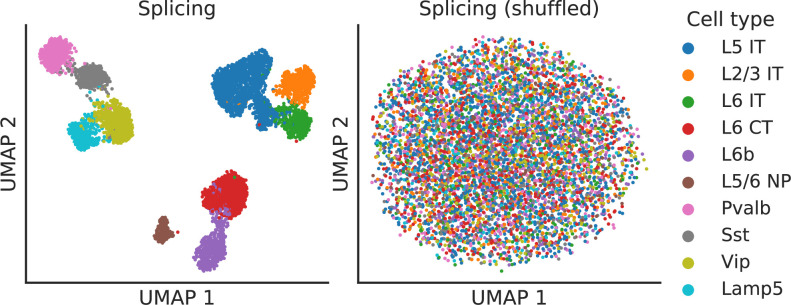
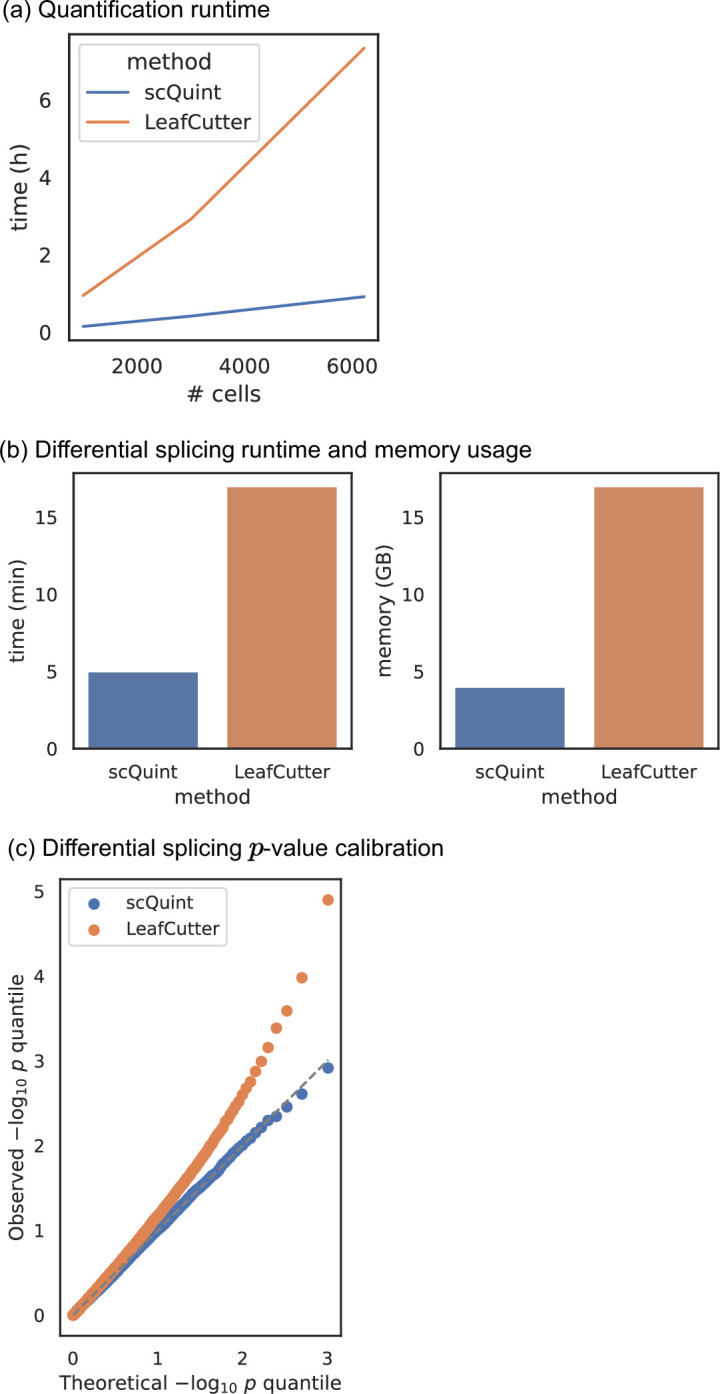
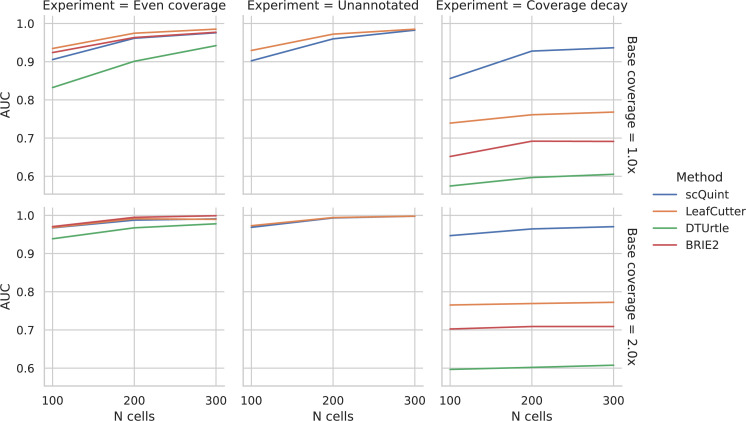
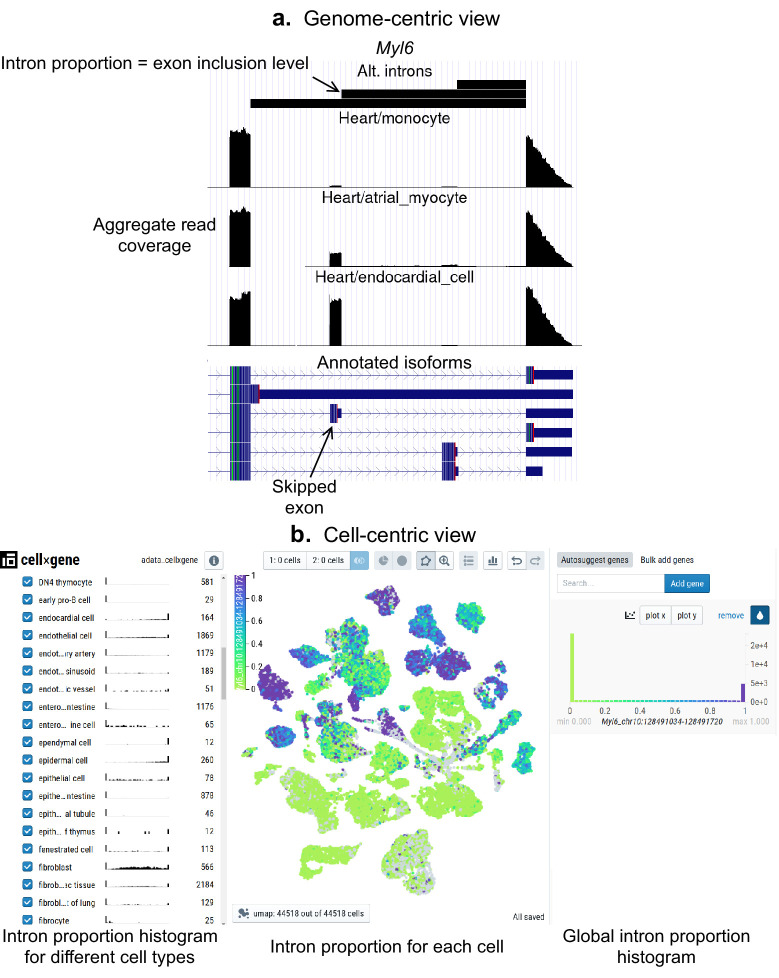
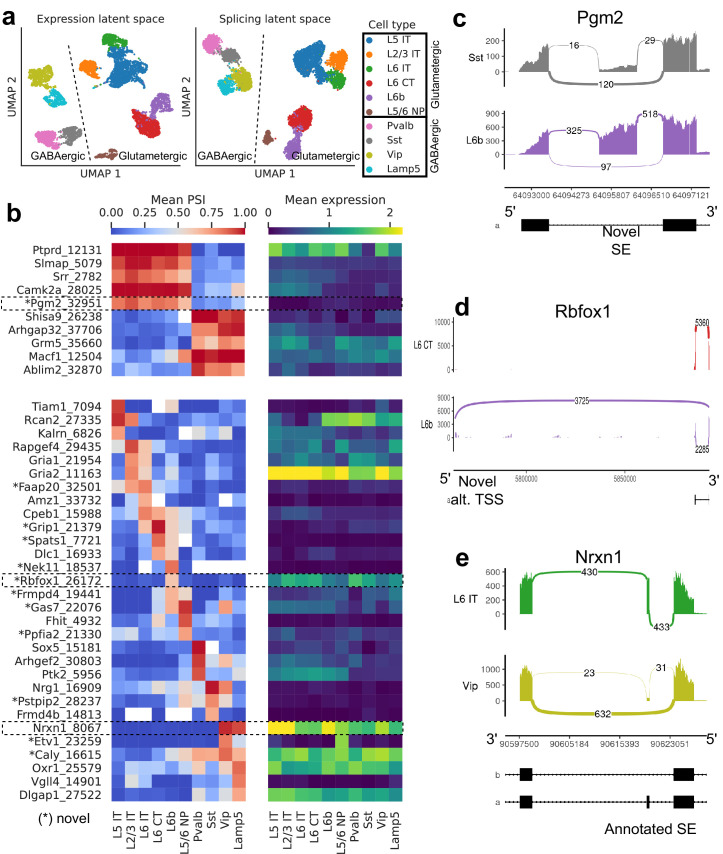
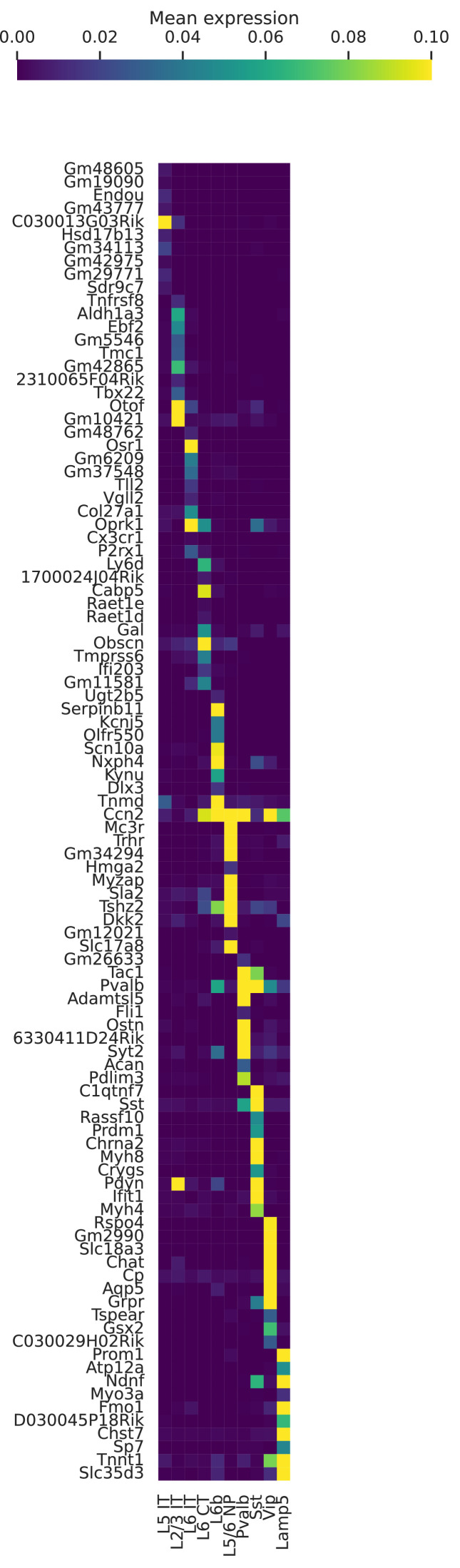
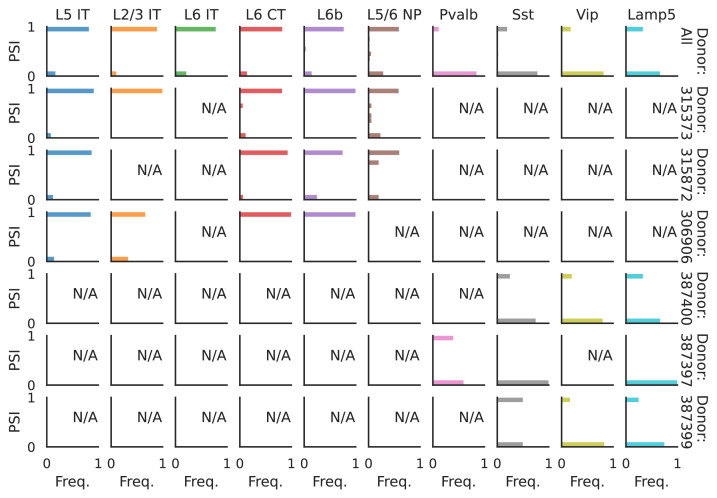
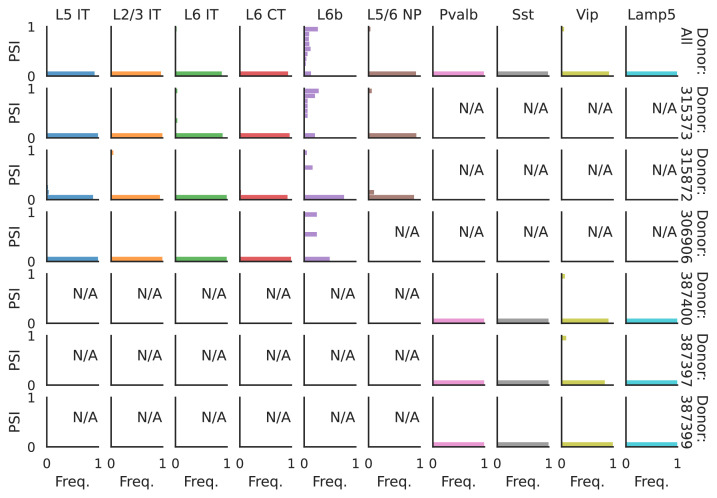
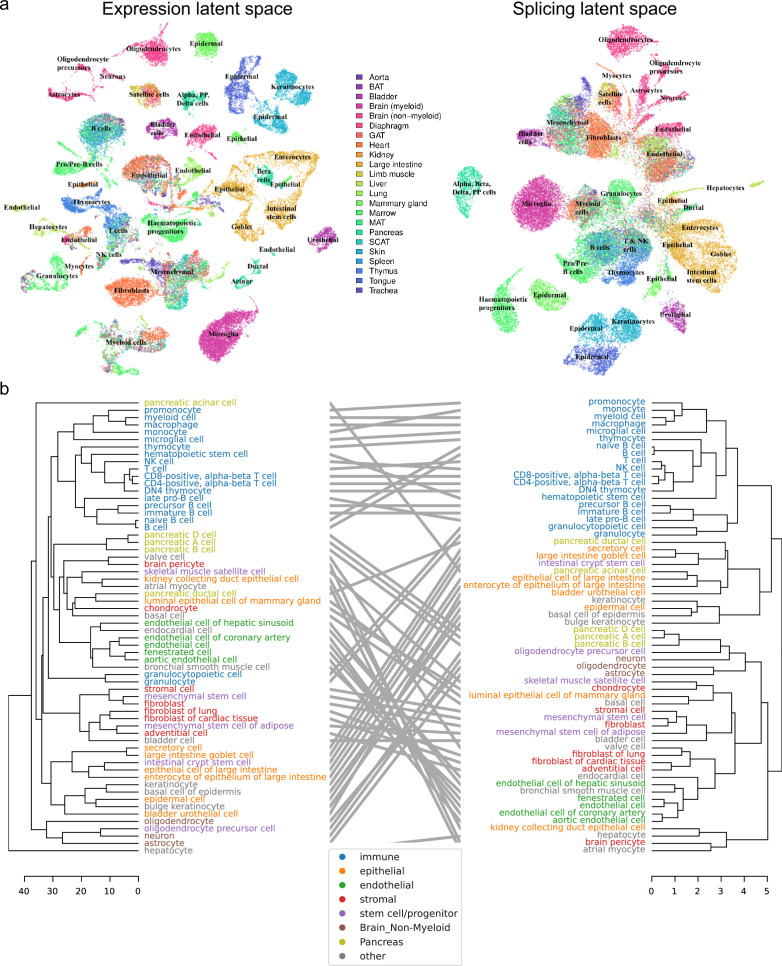
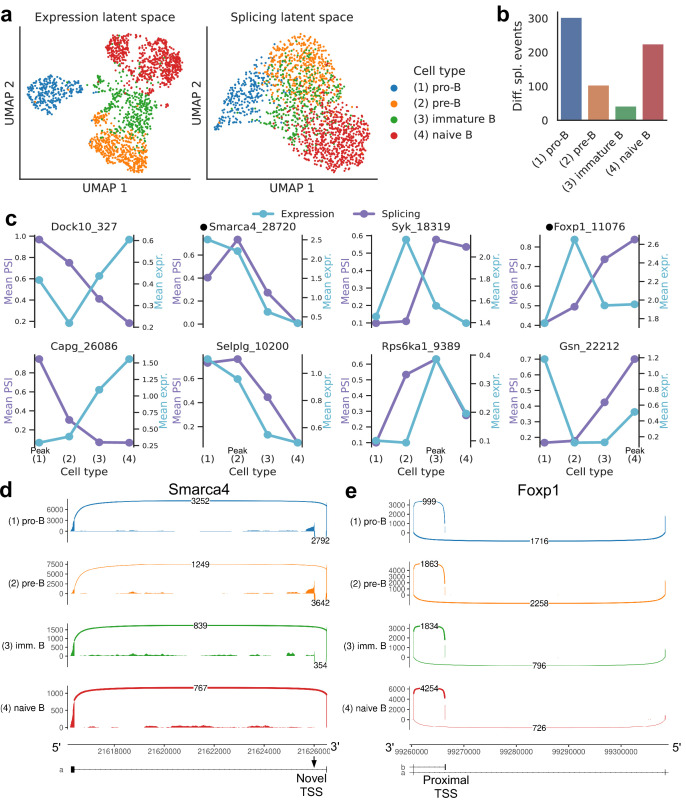
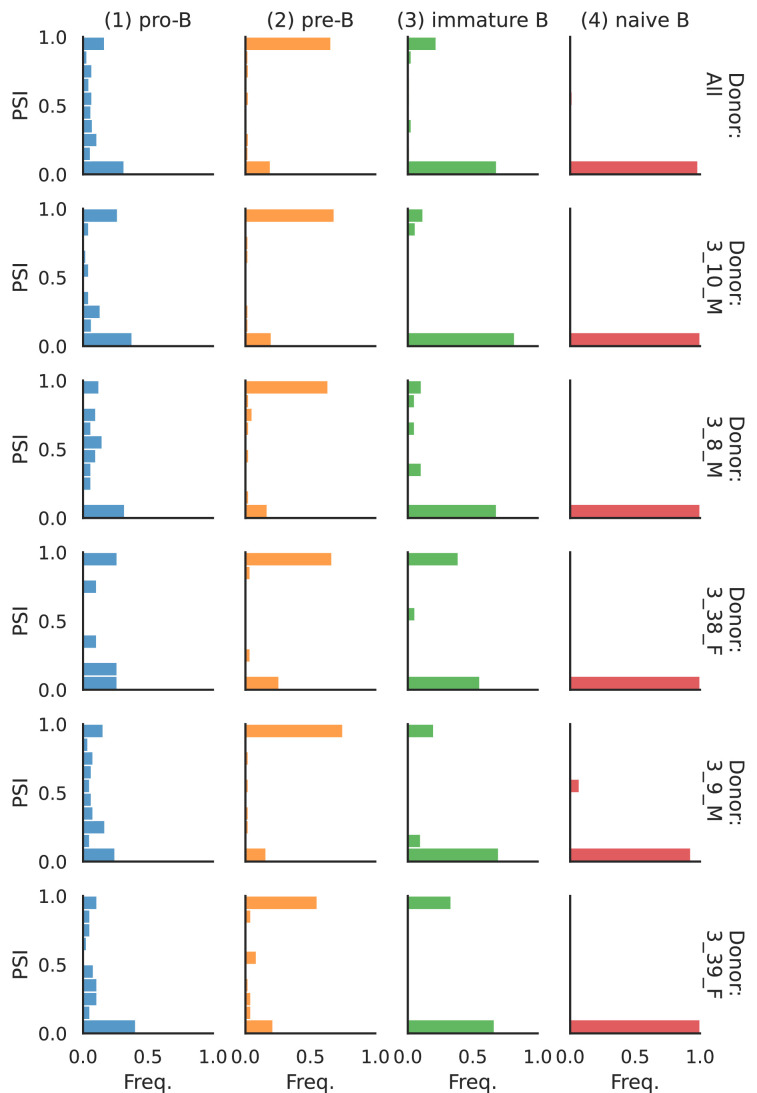
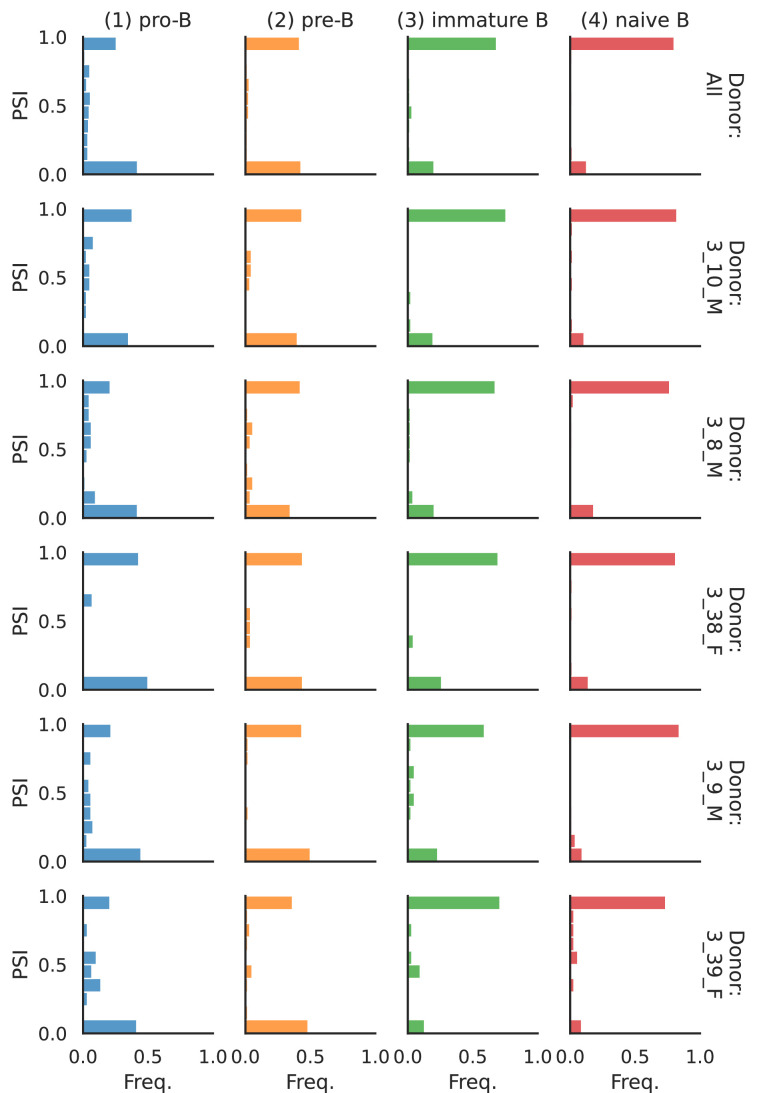
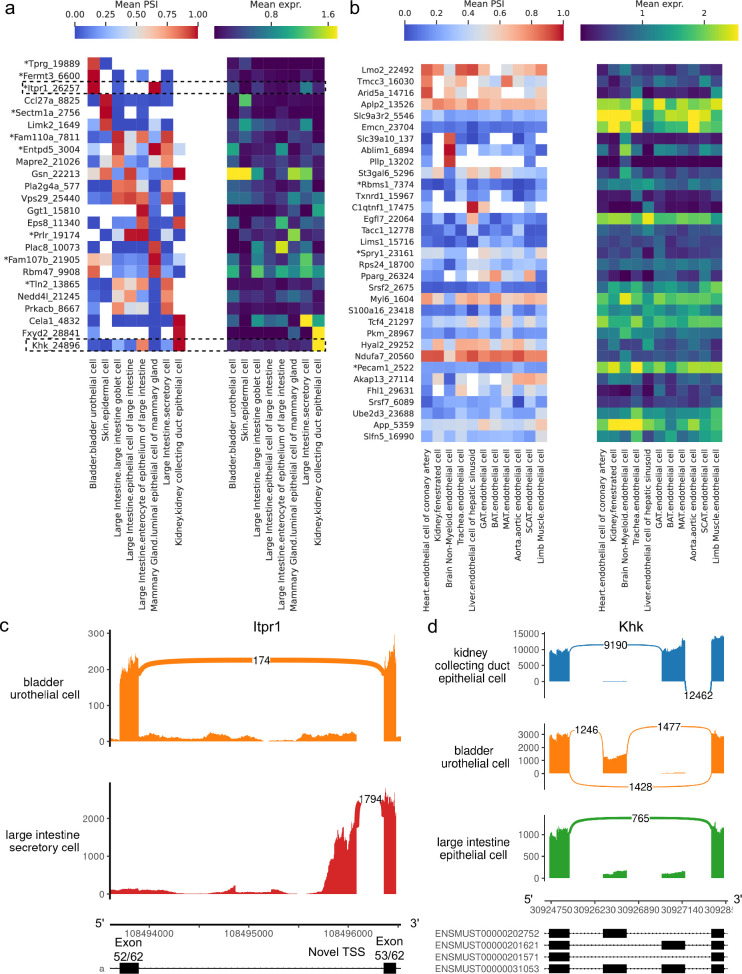
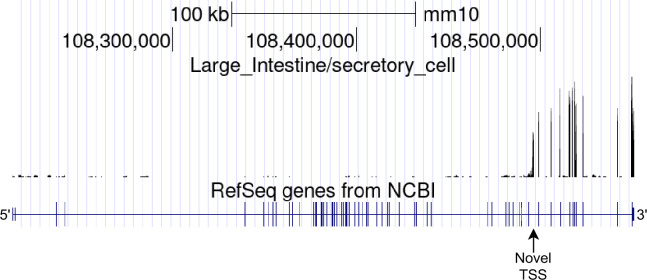
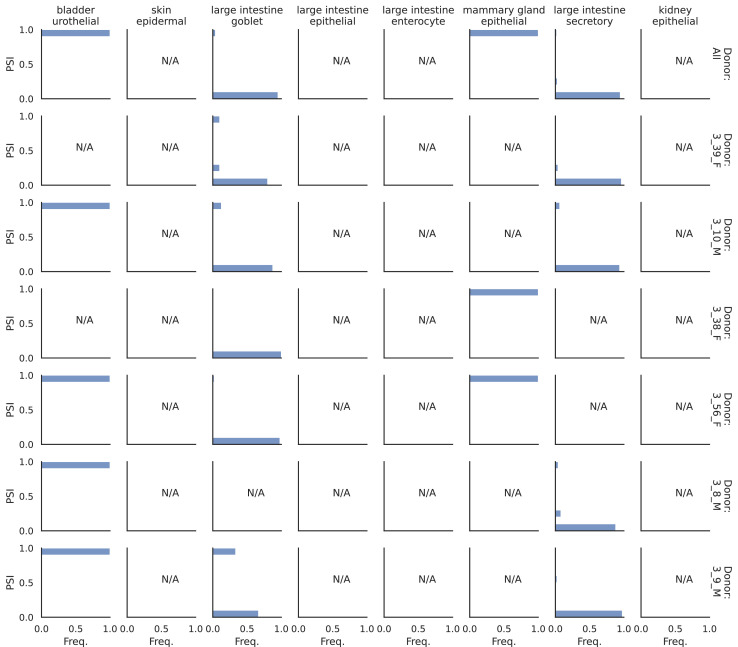
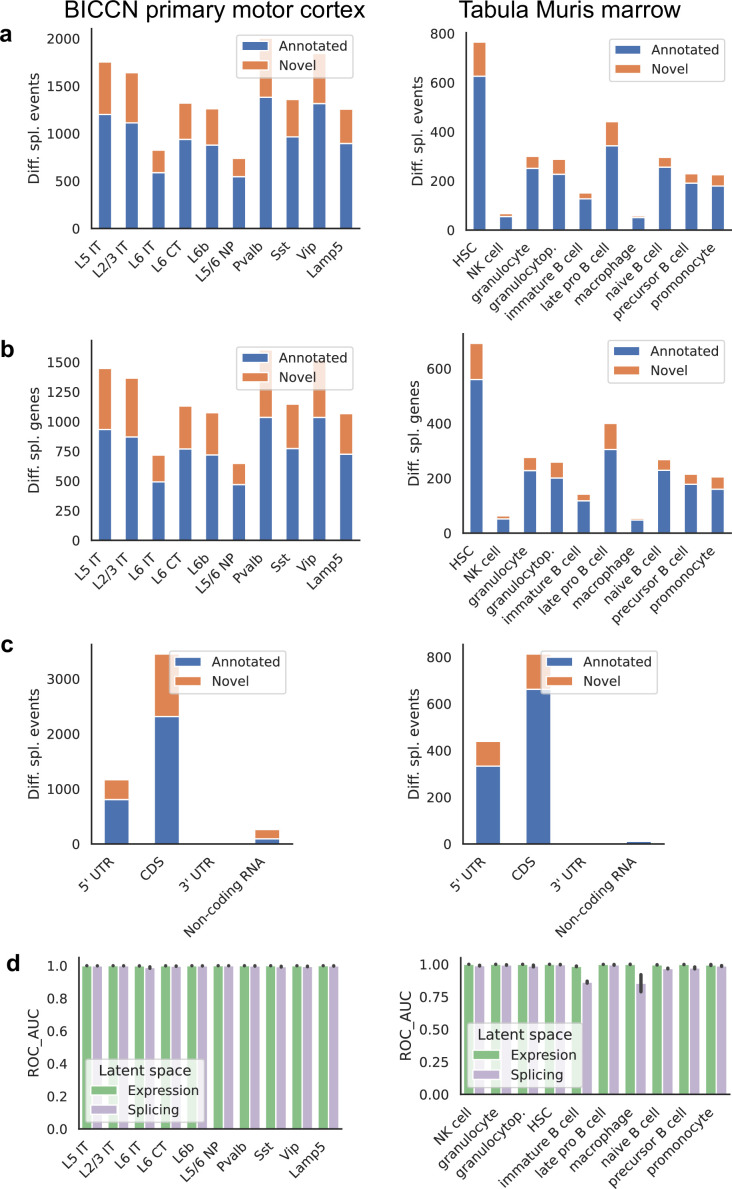
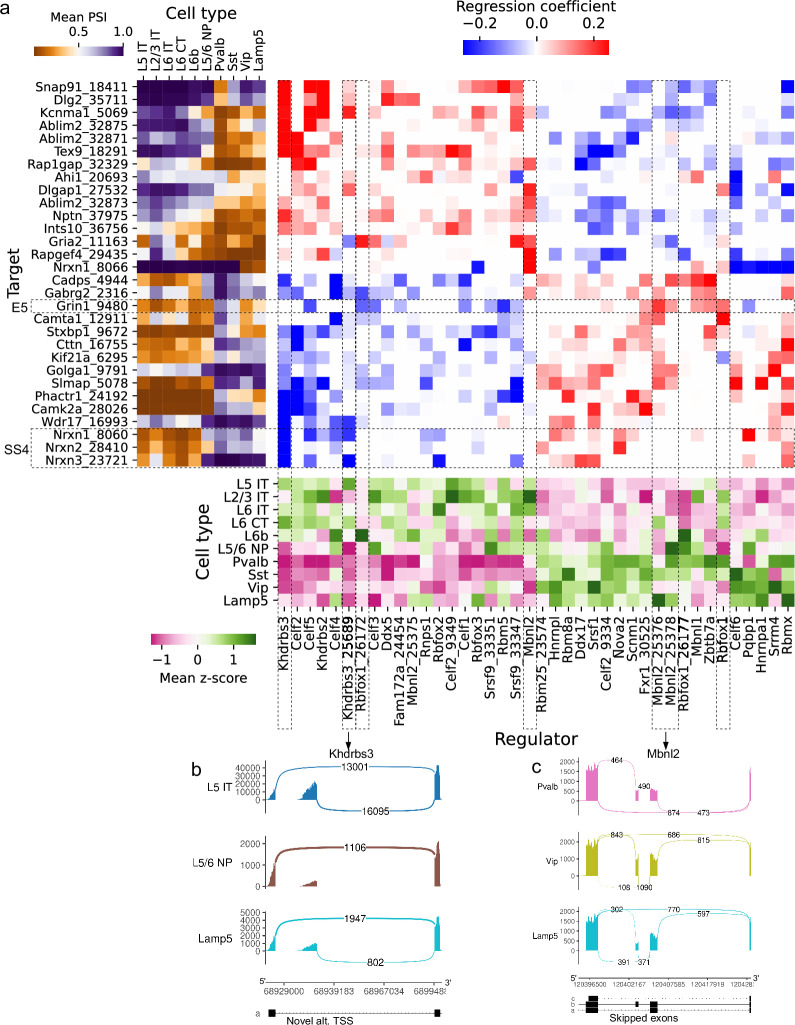
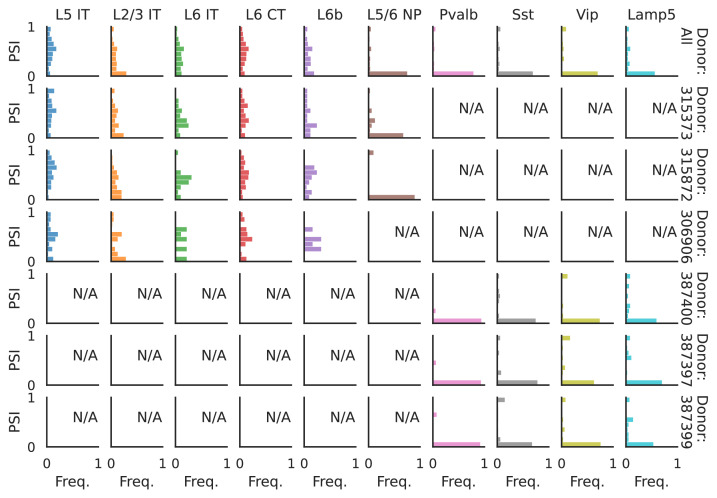
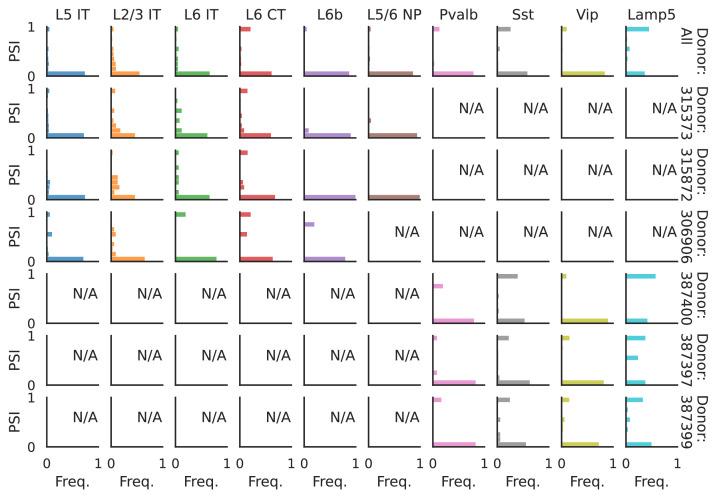
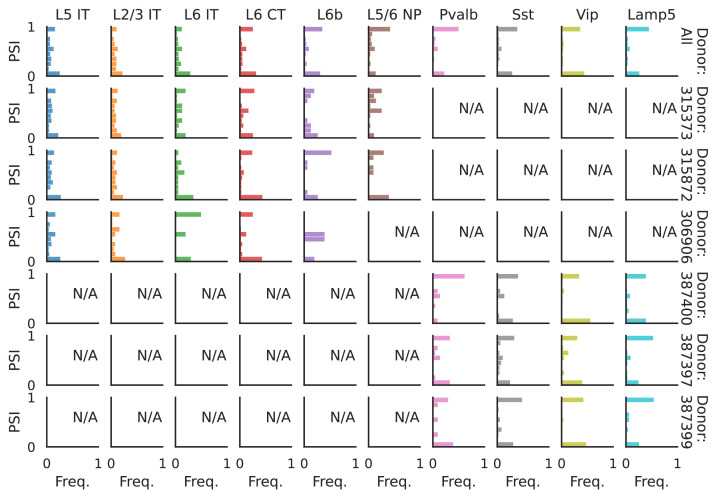
Although alternative splicing is a fundamental and pervasive aspect of gene expression in higher eukaryotes, it is often omitted from single-cell studies due to quantification challenges inherent to commonly used short-read sequencing technologies. Here, we undertake the analysis of alternative splicing across numerous diverse murine cell types from two large-scale single-cell datasets-the Tabula Muris and BRAIN Initiative Cell Census Network-while accounting for understudied technical artifacts and unannotated events. We find strong and general cell-type-specific alternative splicing, complementary to total gene expression but of similar discriminatory value, and identify a large volume of novel splicing events. We specifically highlight splicing variation across different cell types in primary motor cortex neurons, bone marrow B cells, and various epithelial cells, and we show that the implicated transcripts include many genes which do not display total expression differences. To elucidate the regulation of alternative splicing, we build a custom predictive model based on splicing factor activity, recovering several known interactions while generating new hypotheses, including potential regulatory roles for novel alternative splicing events in critical genes like Khdrbs3 and Rbfox1. We make our results available using public interactive browsers to spur further exploration by the community.
Keywords: Tabula Muris; alternative splicing; chromosomes; computational biology; gene expression; mouse; primary motor cortex neurons; single-cell RNA-seq; splicing factors; systems biology.
Plain language summary
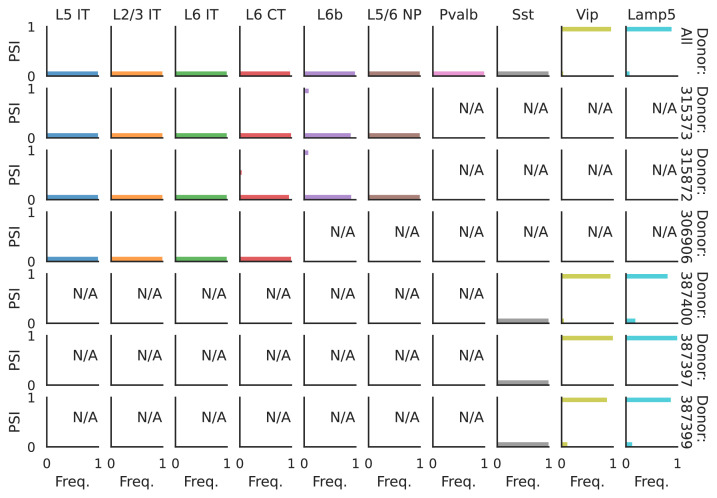
Cells are the basic building blocks of all living things. There are numerous types of cells, and each cell has its own machinery to fulfill a specialised role. Despite their different purposes, most cells contain the same instructions, stored as DNA, on how to assemble the proteins needed to perform their intended functions. Cell types often vary in the frequency that each gene is read, leading to different quantities of proteins produced. Moreover, a process known as alternative splicing enables cells to build multiple proteins from the same gene. It works by joining fragments of a gene’s code in various combinations. The resulting RNA sequences are molecular templates that cells use to assemble proteins. Analysing these RNA sequences reveals which genes are switched on in different tissues of the body, and what proteins are being made. However, despite recent advancements, alternative splicing is rarely studied in single cells because of some sizeable technical challenges. Benegas, Fischer and Song developed a computational toolkit designed to handle the unique challenges of analysing alternative splicing events in single cells. The analysis pipeline, called scQuint, was tested on two large datasets that capture cell-to-cell differences in the brain and other tissues of mice. Nearly all the cell types studied exhibited clear differences in alternative splicing, such that cell types could be distinguished based on their splicing profiles. Intriguing patterns of splicing were highlighted in some immune cells and certain types of neurons. Across cell types, the genes with unique splicing patterns were often not the same as those with unique activity patterns, indicating that gene expression and alternative splicing are two complementary processes. New types of alternative splicing events were also identified. Benegas et al. also developed a statistical model to probe the roles of splicing regulators in different cell types. In summary, the scQuint toolkit overcomes critical technical challenges typically encountered when analysing alternative splicing in single cells. It also reveals new insights about mechanisms of alternative splicing. The results are open access, made available using public interactive browsers, which should spur on other researchers to interrogate how alternative splicing differs in single cells.
© 2022, Benegas et al.
Conflict of interest statement
GB, JF, YS No competing interests declared
Figures


References
Publication types
MeSH terms
Substances
Associated data
- Actions
