

Insight into the ecology of vaginal bacteria through integrative analyses of metagenomic and metatranscriptomic data
- PMID: 35232471
- PMCID: PMC8886902
- DOI: 10.1186/s13059-022-02635-9
Insight into the ecology of vaginal bacteria through integrative analyses of metagenomic and metatranscriptomic data
Abstract
Background: Vaginal bacterial communities dominated by Lactobacillus species are associated with a reduced risk of various adverse health outcomes. However, somewhat unexpectedly, many healthy women have microbiota that are not dominated by lactobacilli. To determine the factors that drive vaginal community composition we characterized the genetic composition and transcriptional activities of vaginal microbiota in healthy women.
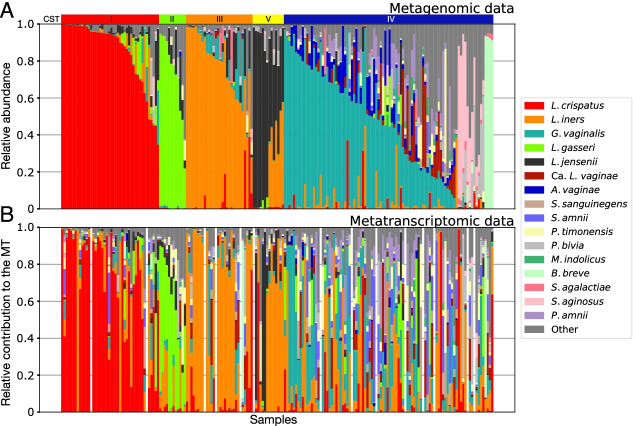
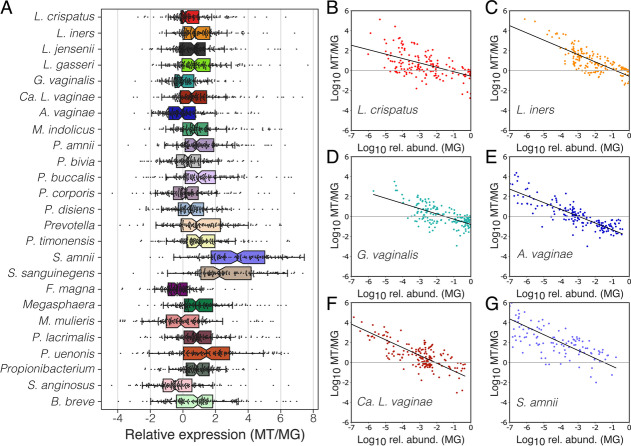
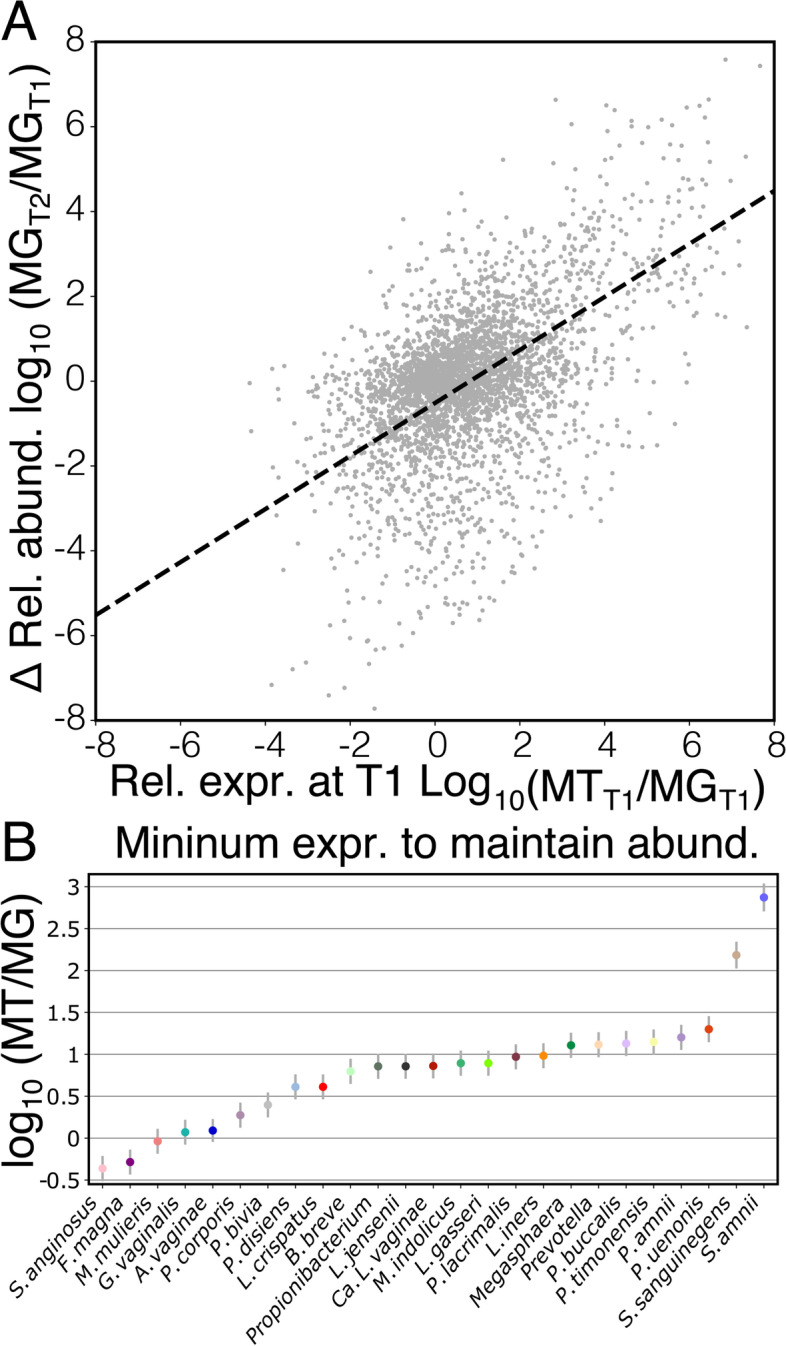
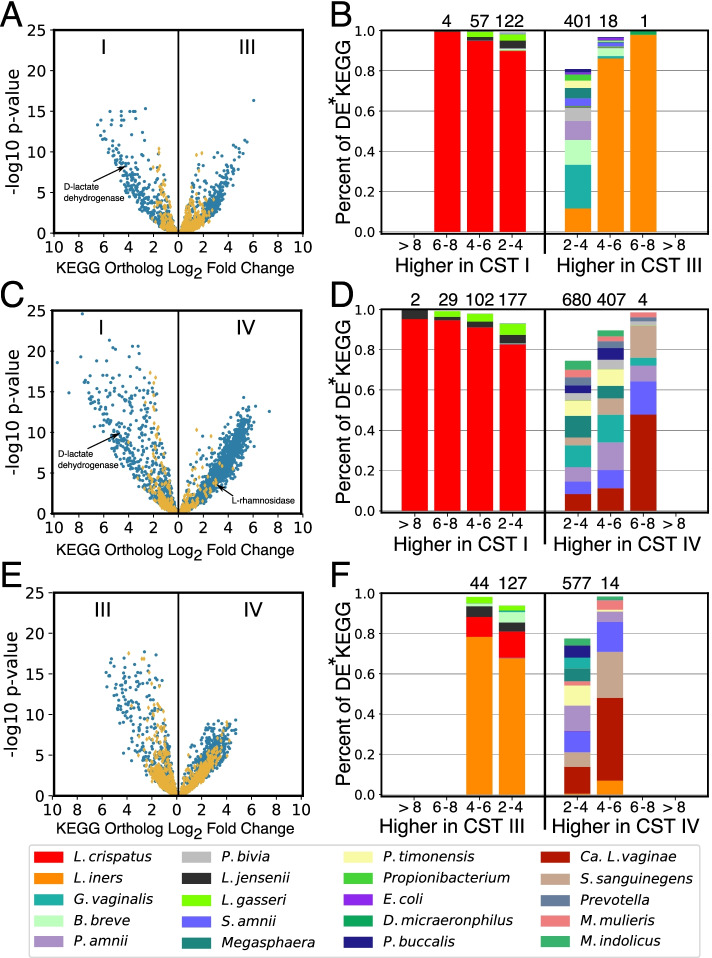
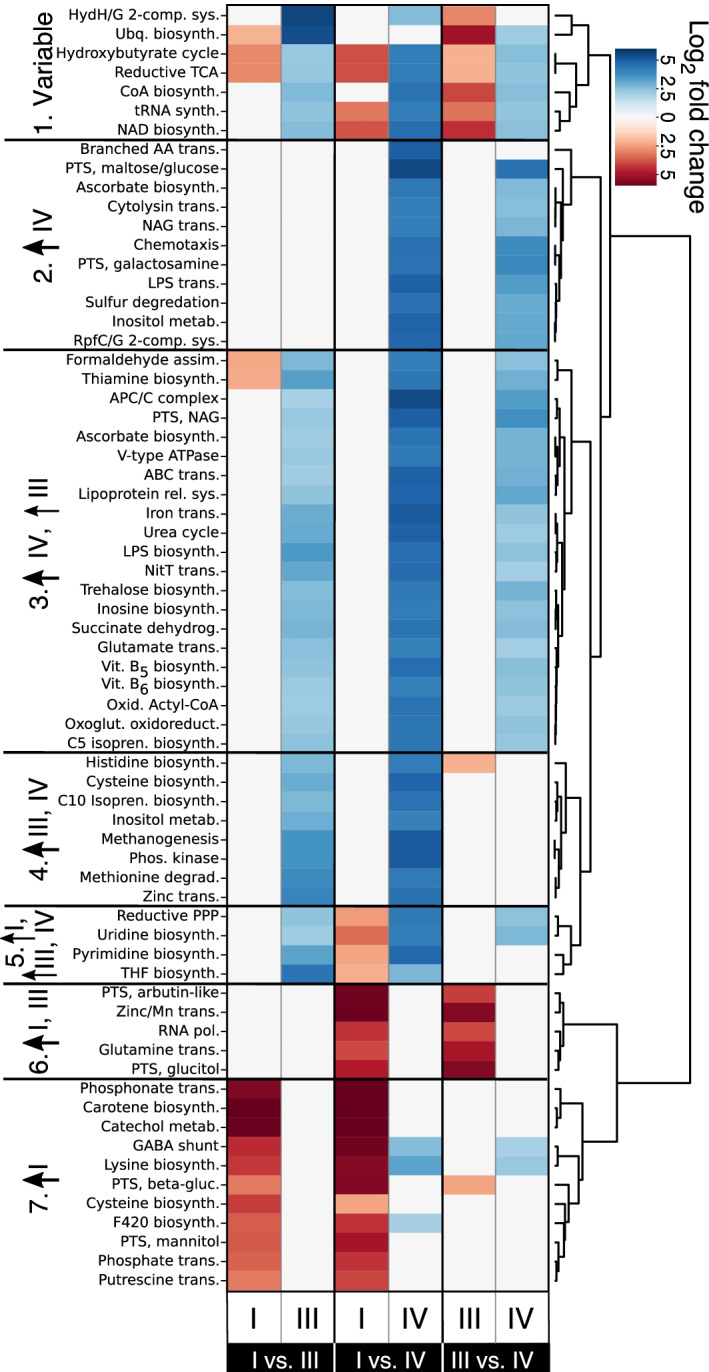
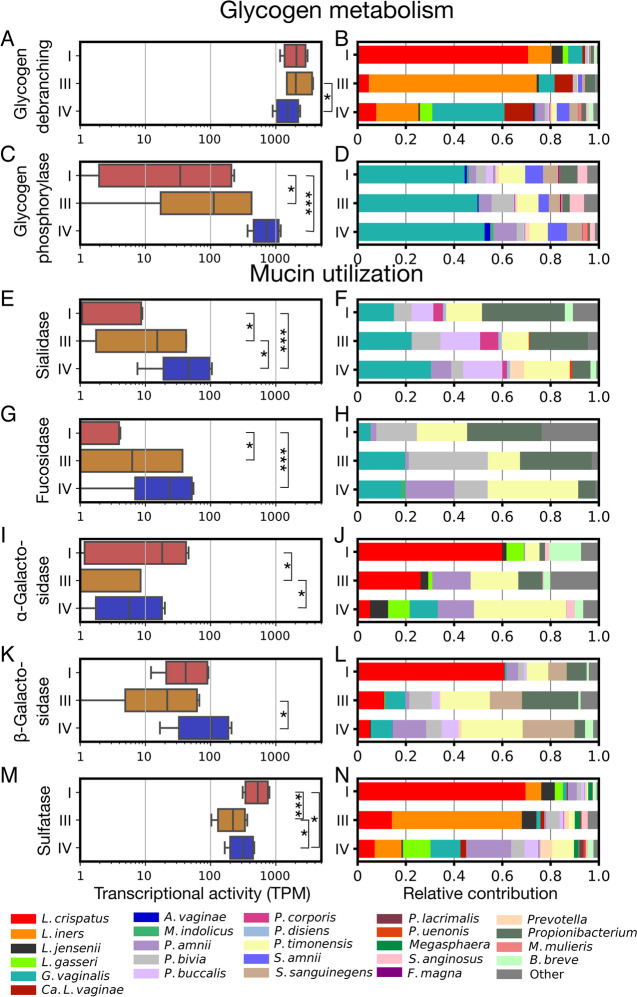
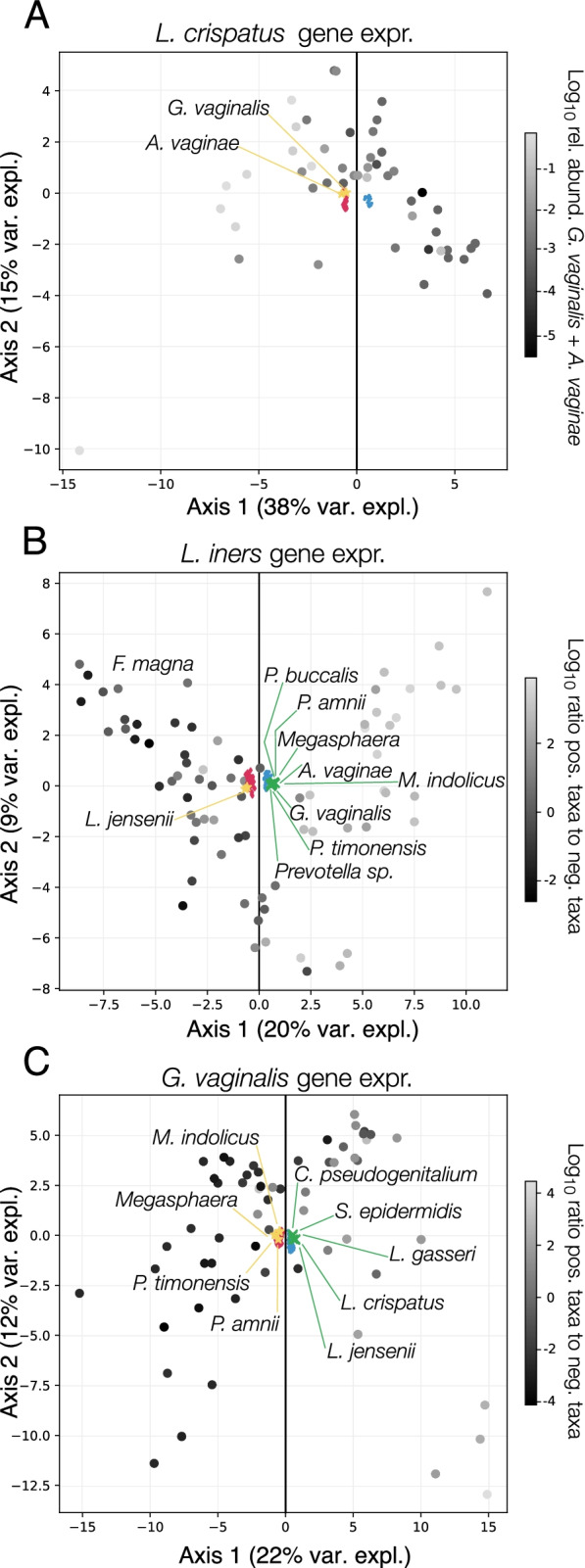
Results: We demonstrate that the abundance of a species is not always indicative of its transcriptional activity and that impending changes in community composition can be predicted from metatranscriptomic data. Functional comparisons highlight differences in the metabolic activities of these communities, notably in their degradation of host produced mucin but not glycogen. Degradation of mucin by communities not dominated by Lactobacillus may play a role in their association with adverse health outcomes. Finally, we show that the transcriptional activities of L. crispatus, L. iners, and Gardnerella vaginalis vary with the taxonomic composition of the communities in which they reside. Notably, L. iners and G. vaginalis both demonstrate lower expression of their cholesterol-dependent cytolysins when co-resident with Lactobacillus spp. and higher expression when co-resident with other facultative and obligate anaerobes. The pathogenic potential of these species may depend on the communities in which they reside and thus could be modulated by interventional strategies.
Conclusions: Our results provide insight to the functional ecology of the vaginal microbiota, demonstrate the diagnostic potential of metatranscriptomic data, and reveal strategies for the management of these ecosystems.
Keywords: Host-microbe interactions; Metagenome; Metatranscriptome; Microbial ecology; Vaginal microbiome.
© 2022. The Author(s).
Conflict of interest statement
J.R. is a cofounder of LUCA Biologics, a biotechnology company focusing on translating microbiome research into live biotherapeutic drugs for women’s health. The other authors declare that they have no competing interests.
Figures
References
-
- Pflughoeft KJ, Versalovic J. Human microbiome in health and disease. Annual Review of Pathology: Mechanisms of Disease: Annual Reviews. 2012. pp. 99–122. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
