

VPipe: an Automated Bioinformatics Platform for Assembly and Management of Viral Next-Generation Sequencing Data
- PMID: 35234489
- PMCID: PMC8941893
- DOI: 10.1128/spectrum.02564-21
VPipe: an Automated Bioinformatics Platform for Assembly and Management of Viral Next-Generation Sequencing Data
Abstract
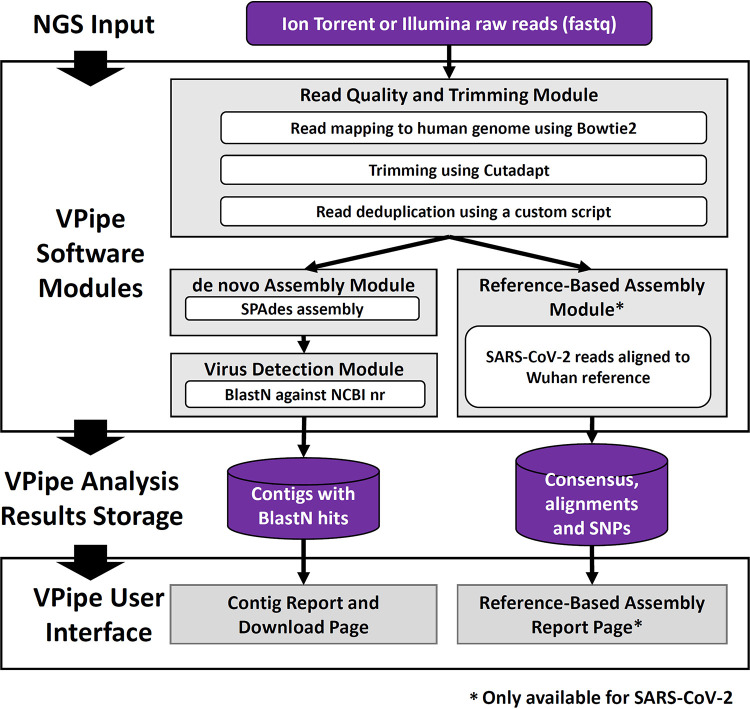
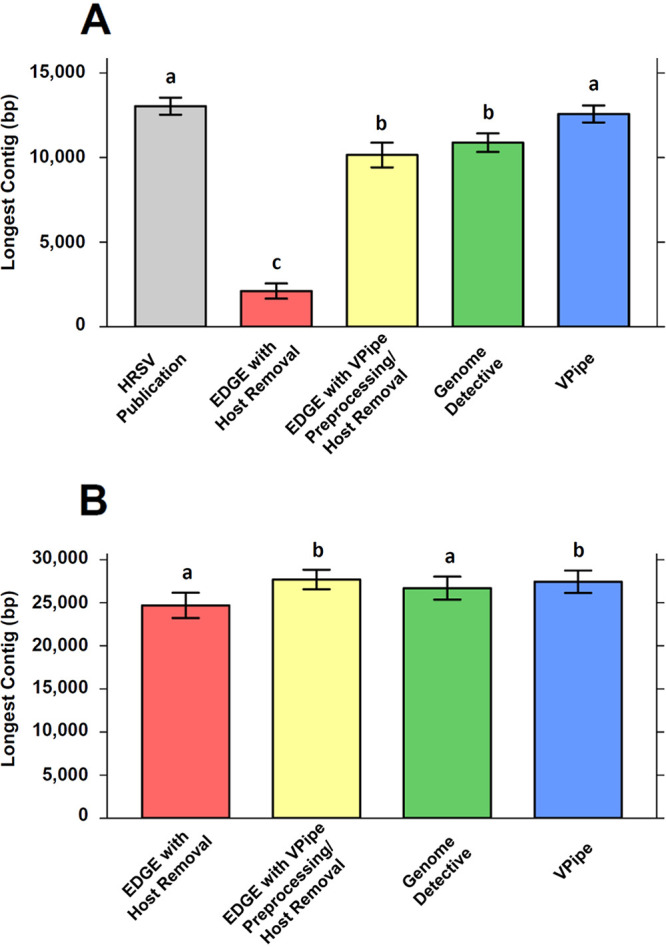
Next-generation sequencing (NGS) is a powerful tool for detecting and investigating viral pathogens; however, analysis and management of the enormous amounts of data generated from these technologies remains a challenge. Here, we present VPipe (the Viral NGS Analysis Pipeline and Data Management System), an automated bioinformatics pipeline optimized for whole-genome assembly of viral sequences and identification of diverse species. VPipe automates the data quality control, assembly, and contig identification steps typically performed when analyzing NGS data. Users access the pipeline through a secure web-based portal, which provides an easy-to-use interface with advanced search capabilities for reviewing results. In addition, VPipe provides a centralized system for storing and analyzing NGS data, eliminating common bottlenecks in bioinformatics analyses for public health laboratories with limited on-site computational infrastructure. The performance of VPipe was validated through the analysis of publicly available NGS data sets for viral pathogens, generating high-quality assemblies for 12 data sets. VPipe also generated assemblies with greater contiguity than similar pipelines for 41 human respiratory syncytial virus isolates and 23 SARS-CoV-2 specimens. IMPORTANCE Computational infrastructure and bioinformatics analysis are bottlenecks in the application of NGS to viral pathogens. As of September 2021, VPipe has been used by the U.S. Centers for Disease Control and Prevention (CDC) and 12 state public health laboratories to characterize >17,500 and 1,500 clinical specimens and isolates, respectively. VPipe automates genome assembly for a wide range of viruses, including high-consequence pathogens such as SARS-CoV-2. Such automated functionality expedites public health responses to viral outbreaks and pathogen surveillance.
Keywords: automated bioinformatics pipeline; infectious disease surveillance; next-generation sequencing (NGS); viral molecular detection.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Maljkovic Berry I, Melendrez MC, Bishop-Lilly KA, Rutvisuttinunt W, Pollett S, Talundzic E, Morton L, Jarman RG. 2020. Next generation sequencing and bioinformatics methodologies for infectious disease research and public health: approaches, applications, and considerations for development of laboratory capacity. J Infect Dis 221:S292–S307. doi: 10.1093/infdis/jiz286. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
