

Secondary structural ensembles of the SARS-CoV-2 RNA genome in infected cells
- PMID: 35236847
- PMCID: PMC8891300
- DOI: 10.1038/s41467-022-28603-2
Secondary structural ensembles of the SARS-CoV-2 RNA genome in infected cells
Abstract
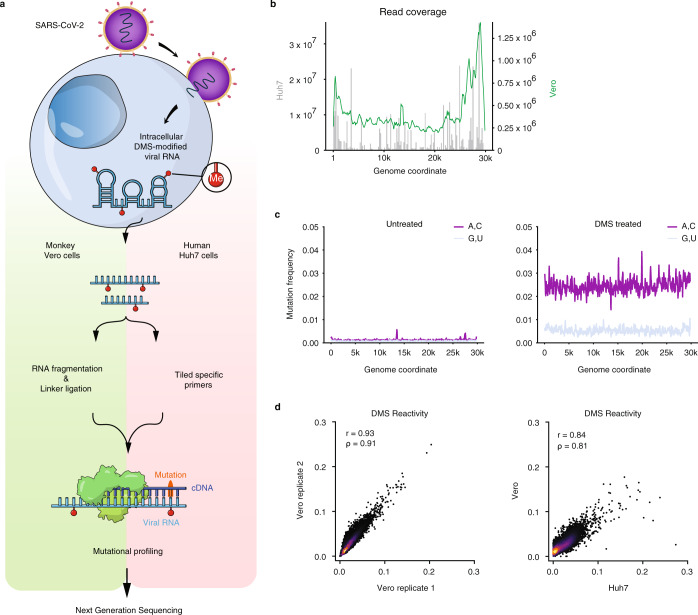
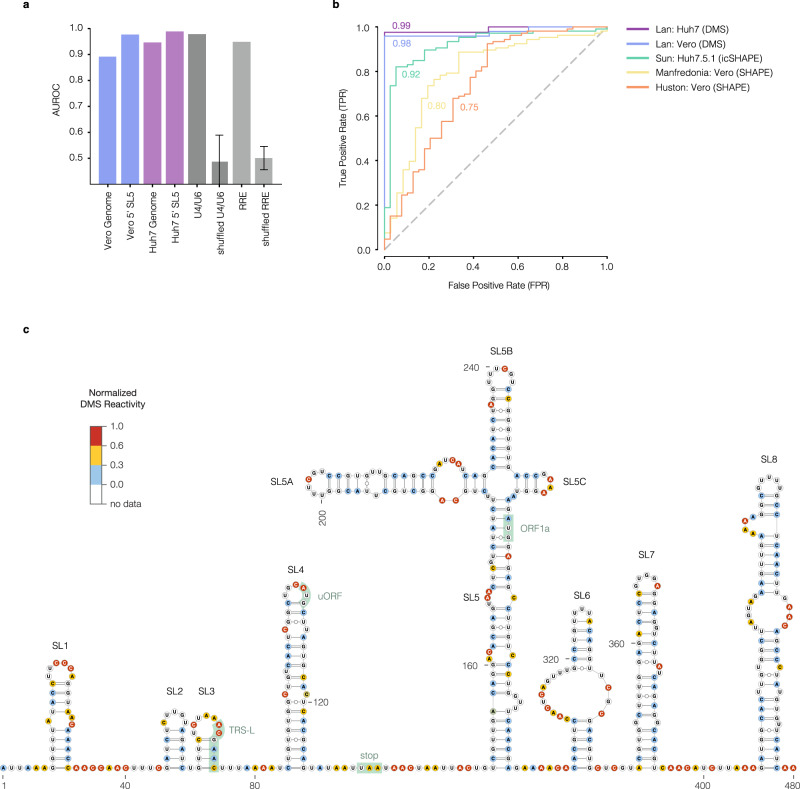
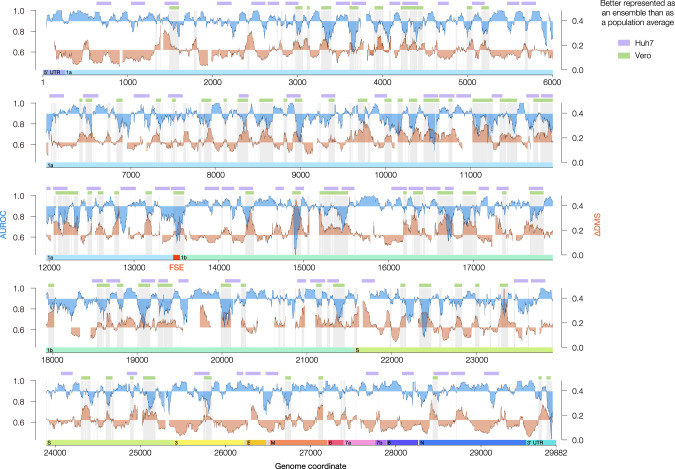
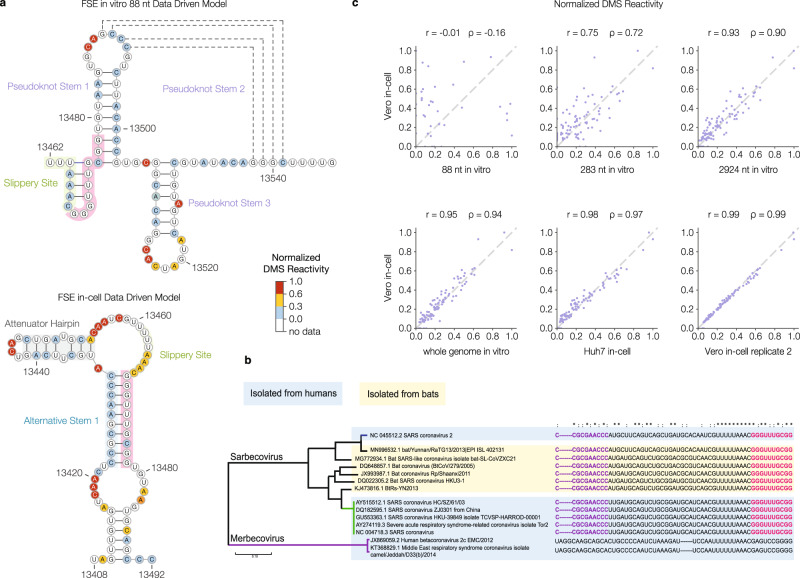
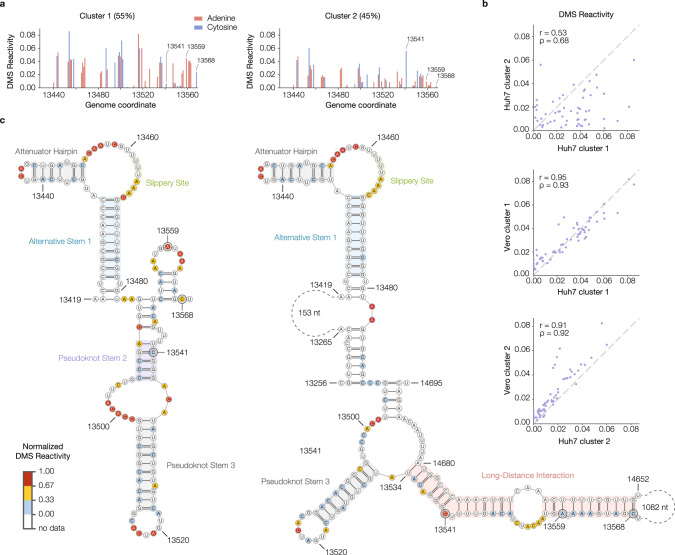
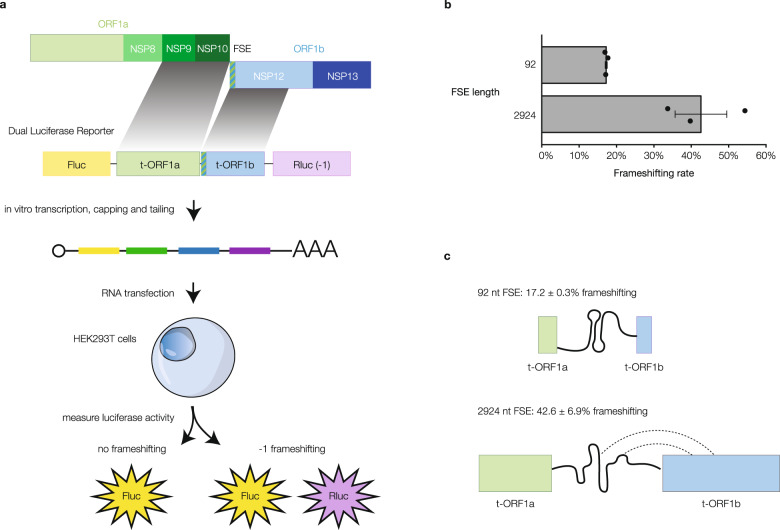
SARS-CoV-2 is a betacoronavirus with a single-stranded, positive-sense, 30-kilobase RNA genome responsible for the ongoing COVID-19 pandemic. Although population average structure models of the genome were recently reported, there is little experimental data on native structural ensembles, and most structures lack functional characterization. Here we report secondary structure heterogeneity of the entire SARS-CoV-2 genome in two lines of infected cells at single nucleotide resolution. Our results reveal alternative RNA conformations across the genome and at the critical frameshifting stimulation element (FSE) that are drastically different from prevailing population average models. Importantly, we find that this structural ensemble promotes frameshifting rates much higher than the canonical minimal FSE and similar to ribosome profiling studies. Our results highlight the value of studying RNA in its full length and cellular context. The genomic structures detailed here lay groundwork for coronavirus RNA biology and will guide the design of SARS-CoV-2 RNA-based therapeutics.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
