

Two zinc finger proteins with functions in m6A writing interact with HAKAI
- PMID: 35236848
- PMCID: PMC8891334
- DOI: 10.1038/s41467-022-28753-3
Two zinc finger proteins with functions in m6A writing interact with HAKAI
Abstract
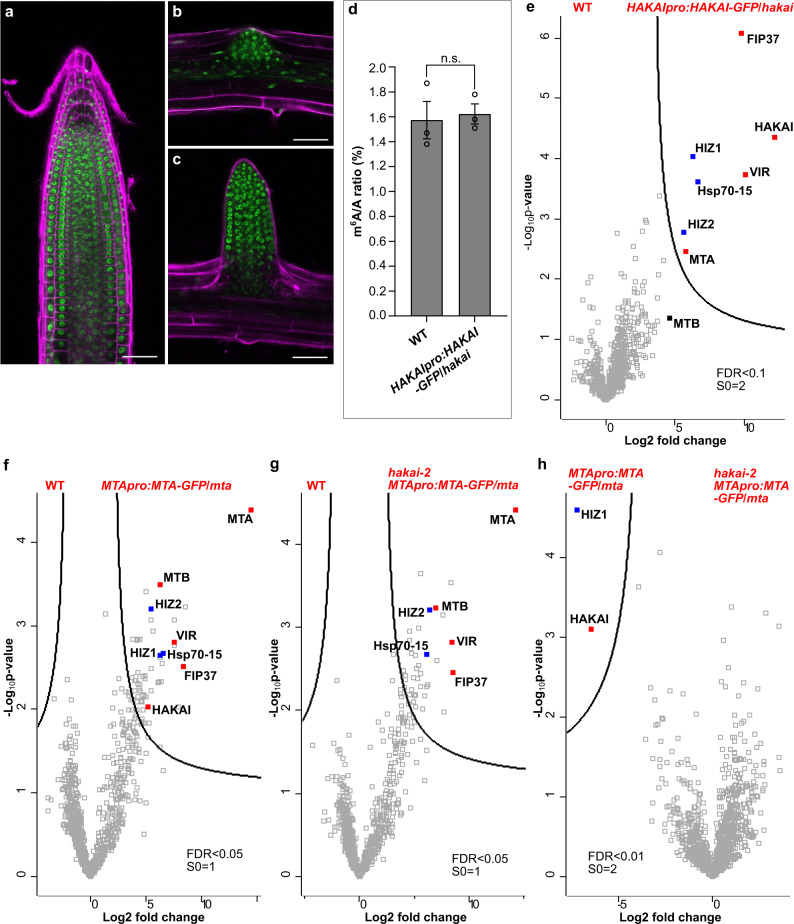
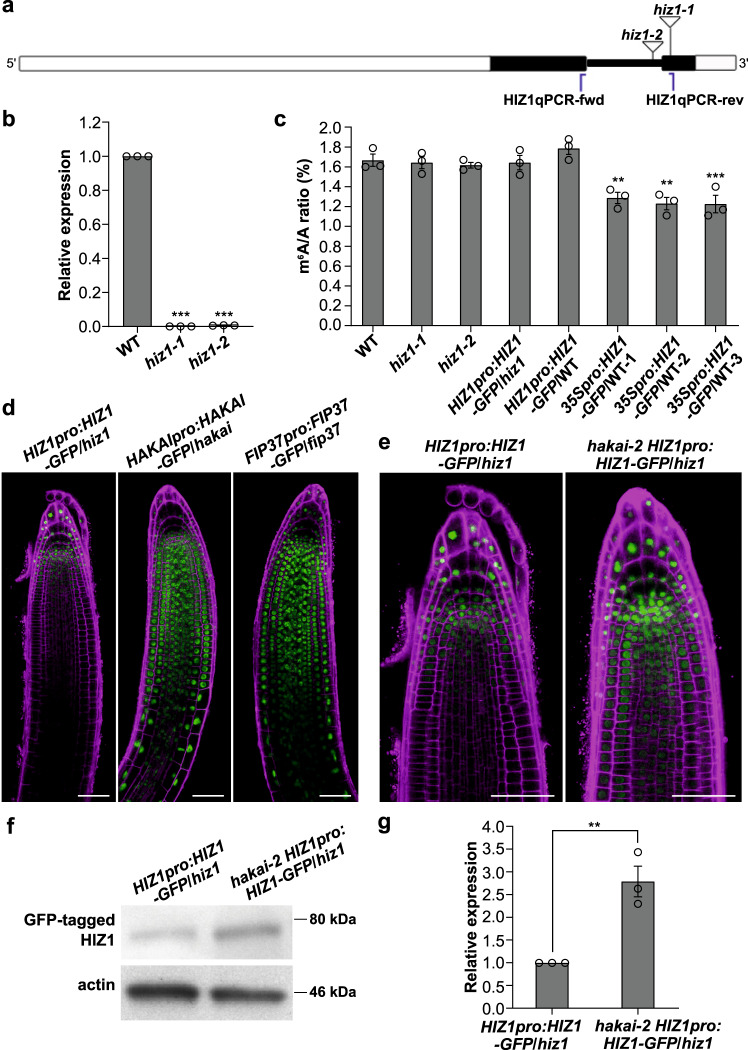
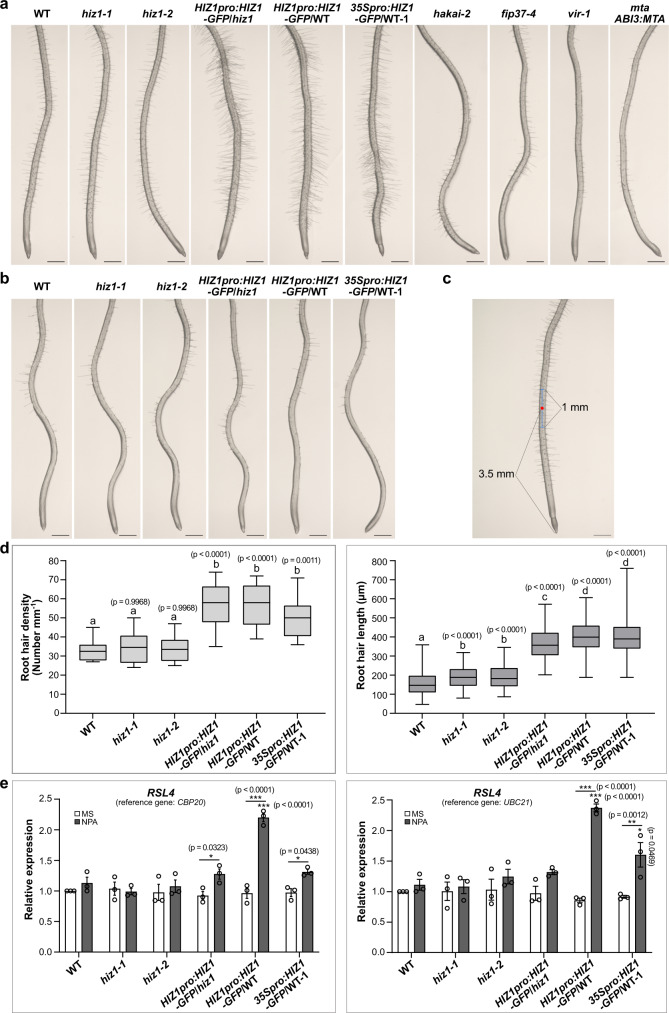
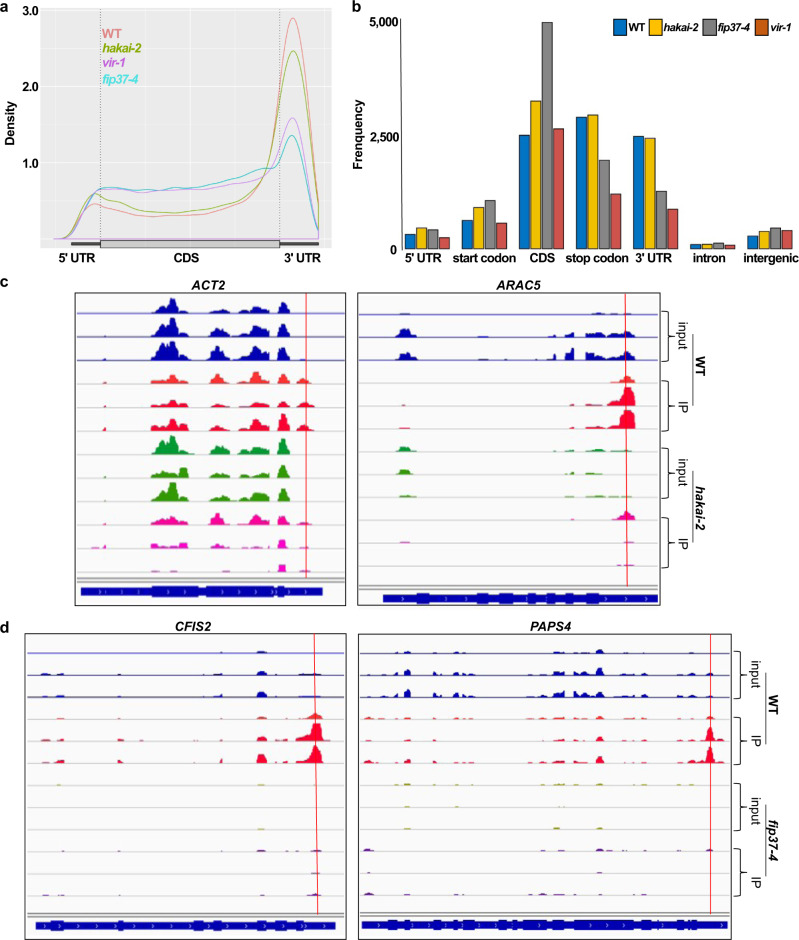
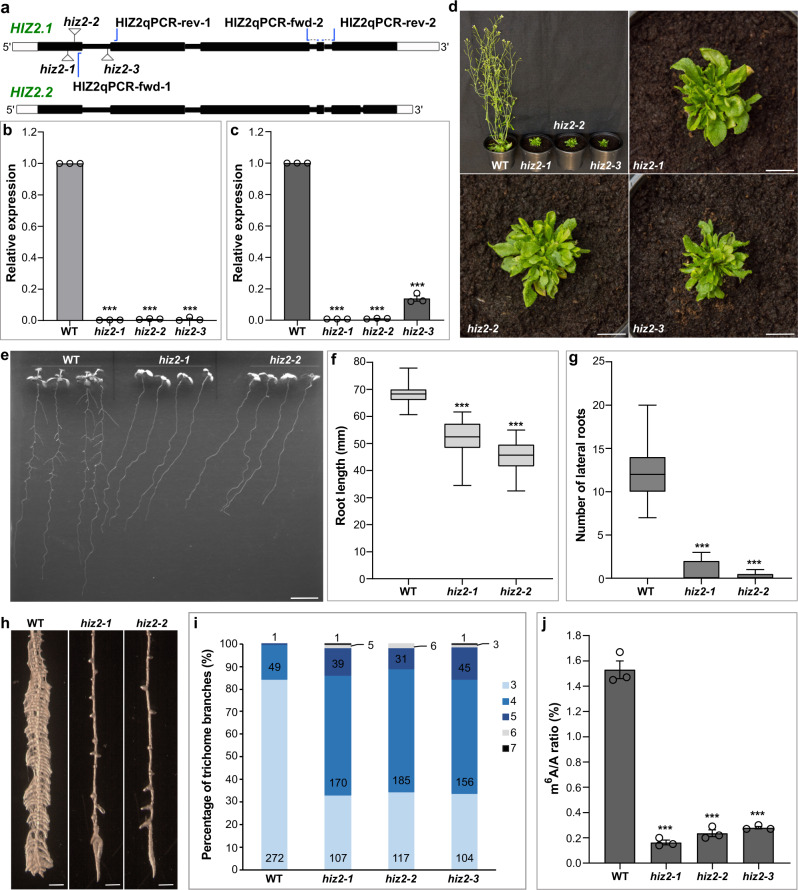
The methyltransferase complex (m6A writer), which catalyzes the deposition of N6-methyladenosine (m6A) in mRNAs, is highly conserved across most eukaryotic organisms, but its components and interactions between them are still far from fully understood. Here, using in vivo interaction proteomics, two HAKAI-interacting zinc finger proteins, HIZ1 and HIZ2, are discovered as components of the Arabidopsis m6A writer complex. HAKAI is required for the interaction between HIZ1 and MTA (mRNA adenosine methylase A). Whilst HIZ1 knockout plants have normal levels of m6A, plants in which it is overexpressed show reduced methylation and decreased lateral root formation. Mutant plants lacking HIZ2 are viable but have an 85% reduction in m6A abundance and show severe developmental defects. Our findings suggest that HIZ2 is likely the plant equivalent of ZC3H13 (Flacc) of the metazoan m6A-METTL Associated Complex.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Bokar JA, Rath-Shambaugh ME, Ludwiczak R, Narayan P, Rottman F. Characterization and partial purification of mRNA N6-adenosine methyltransferase from HeLa cell nuclei. Internal mRNA methylation requires a multisubunit complex. J. Biol. Chem. 1994;269:17697–17704. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
