

Molecular hallmarks of heterochronic parabiosis at single-cell resolution
- PMID: 35236985
- PMCID: PMC9387403
- DOI: 10.1038/s41586-022-04461-2
Molecular hallmarks of heterochronic parabiosis at single-cell resolution
Abstract
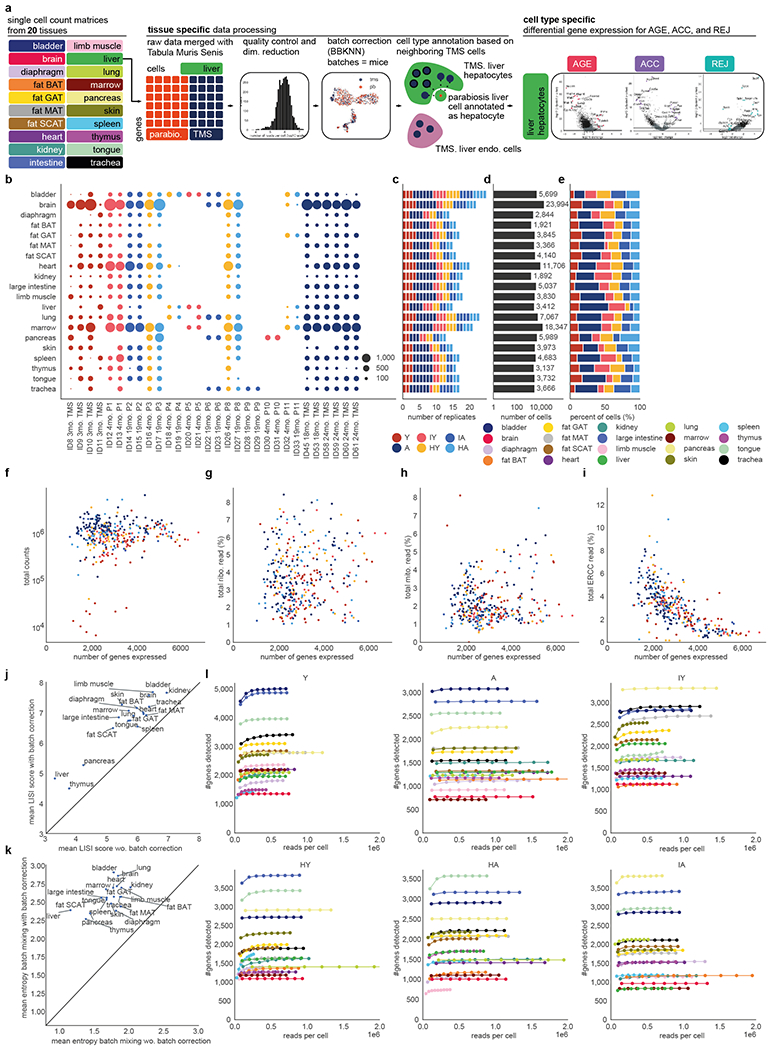
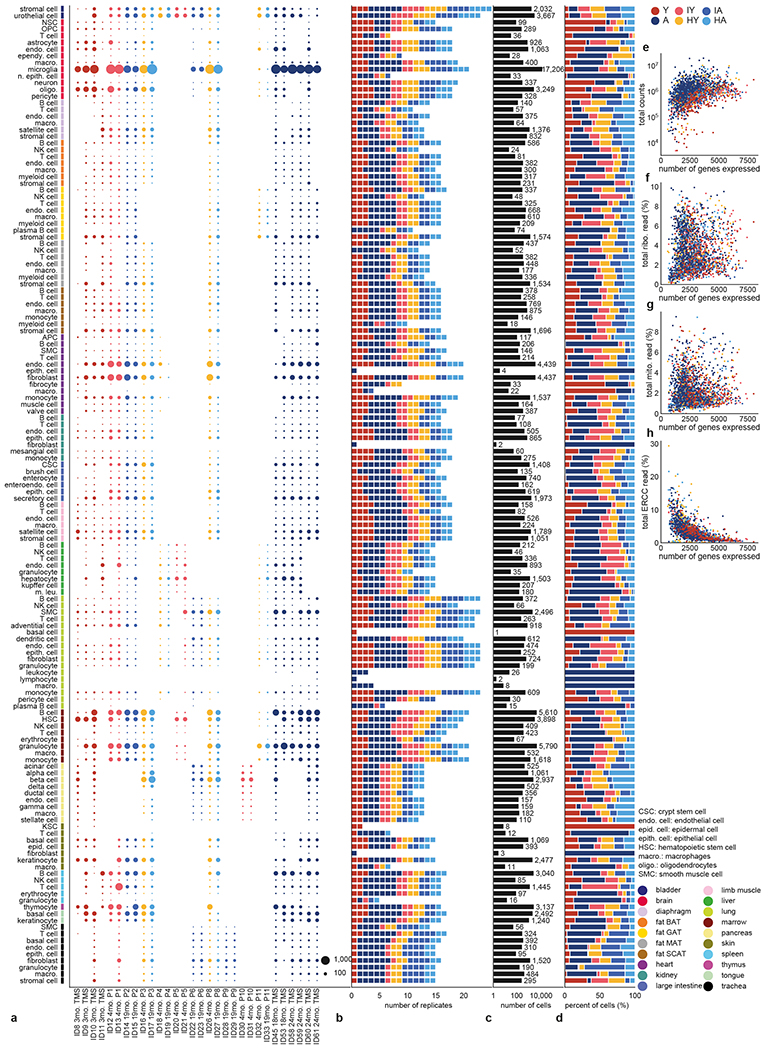
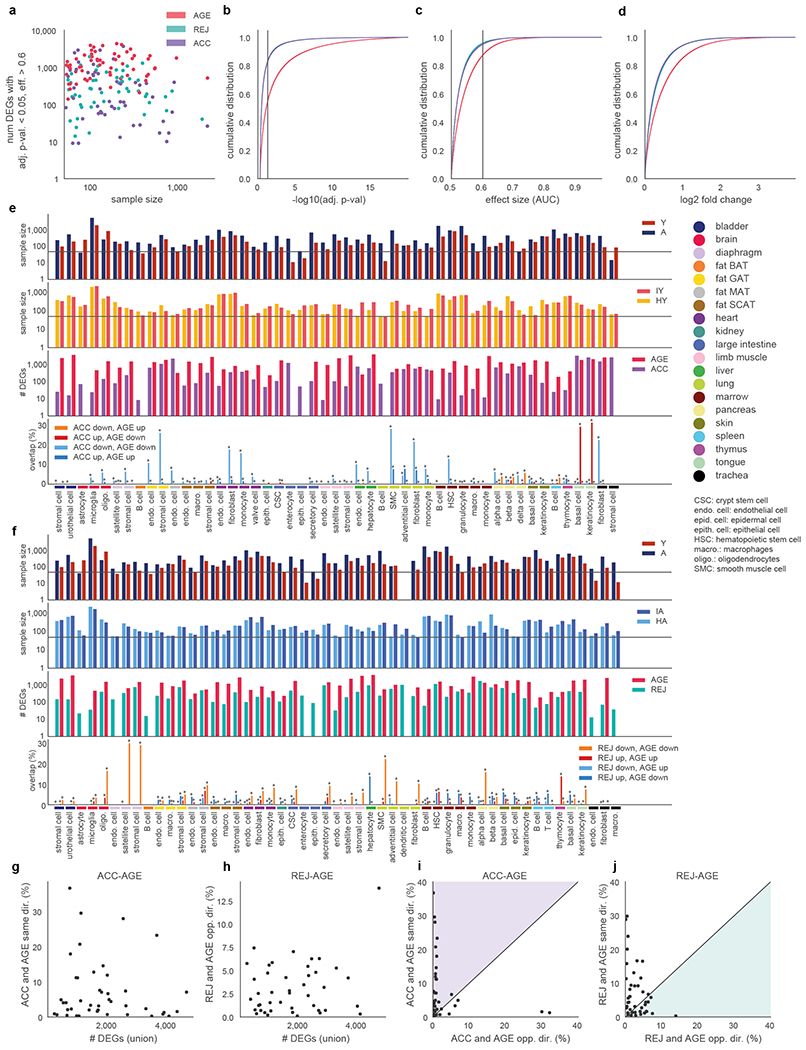
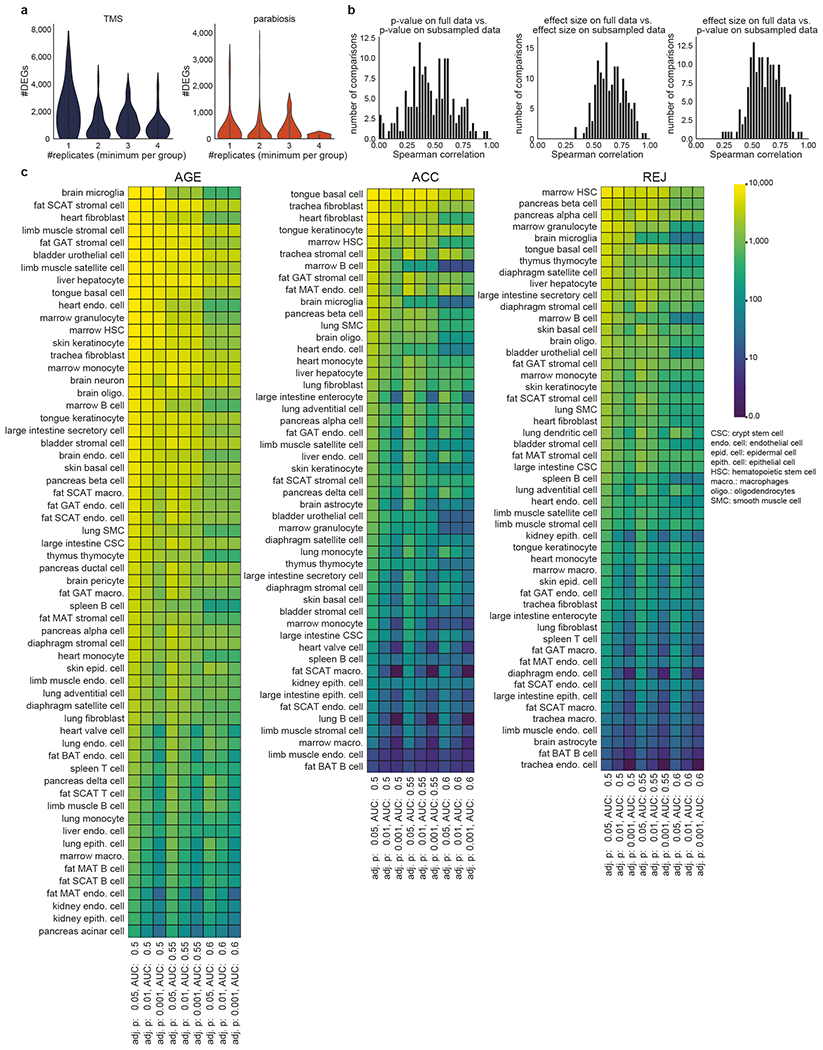
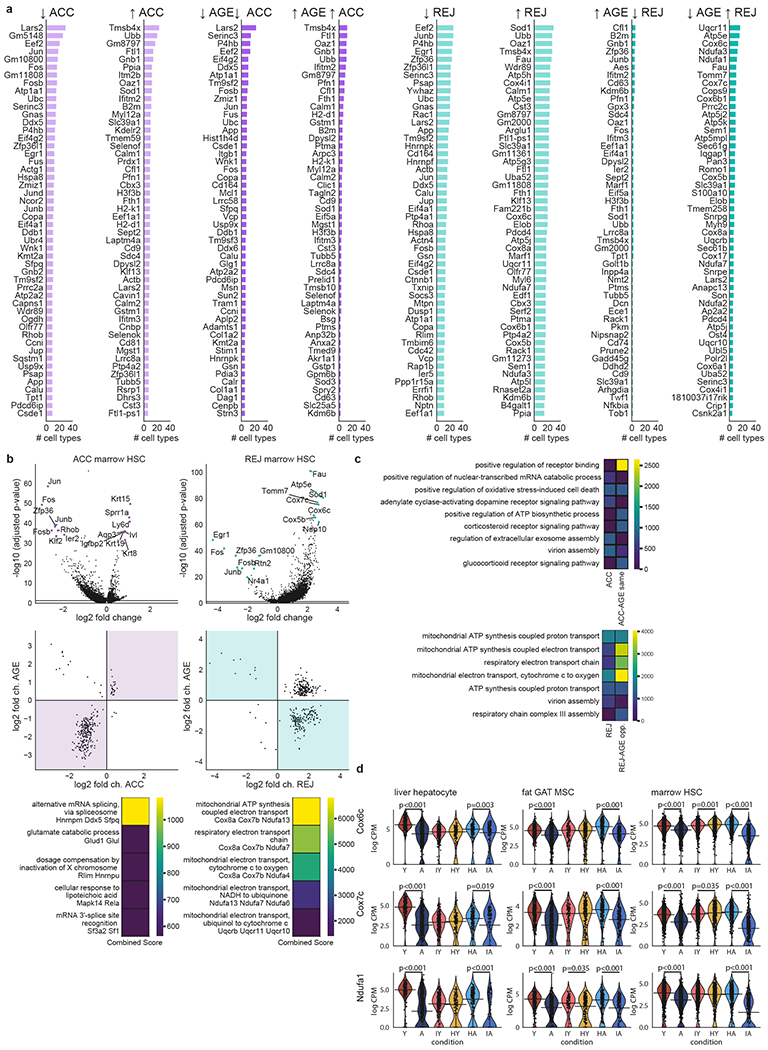
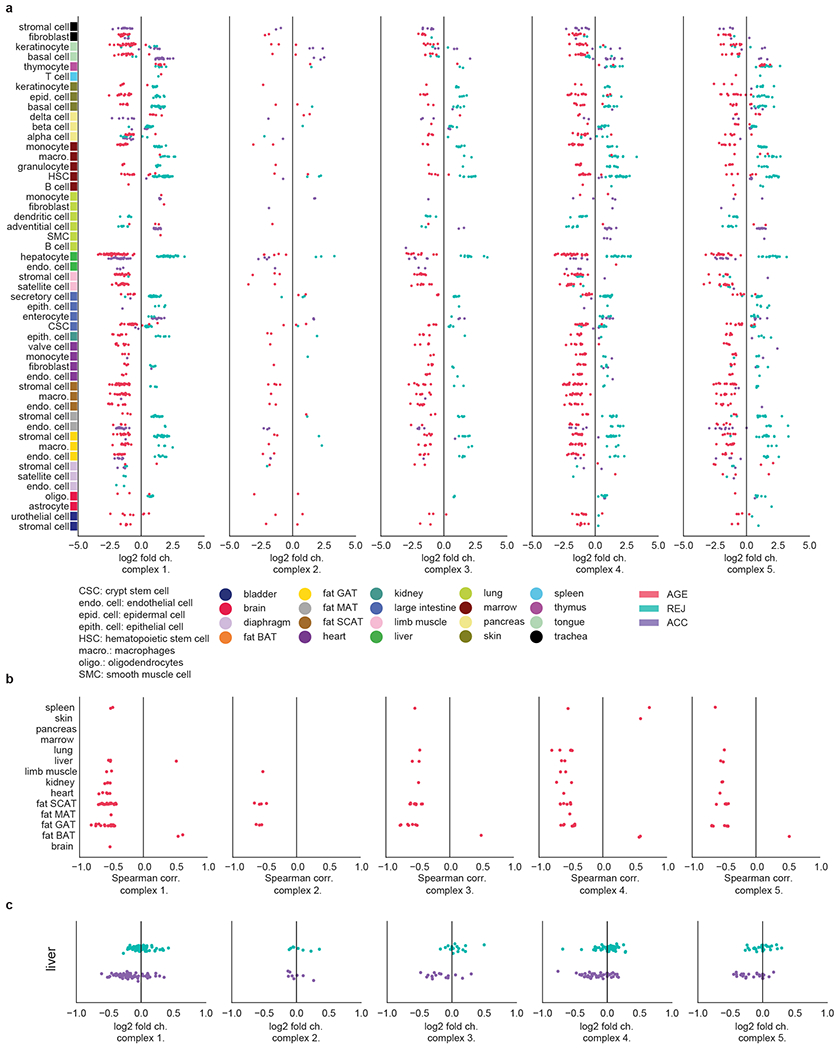
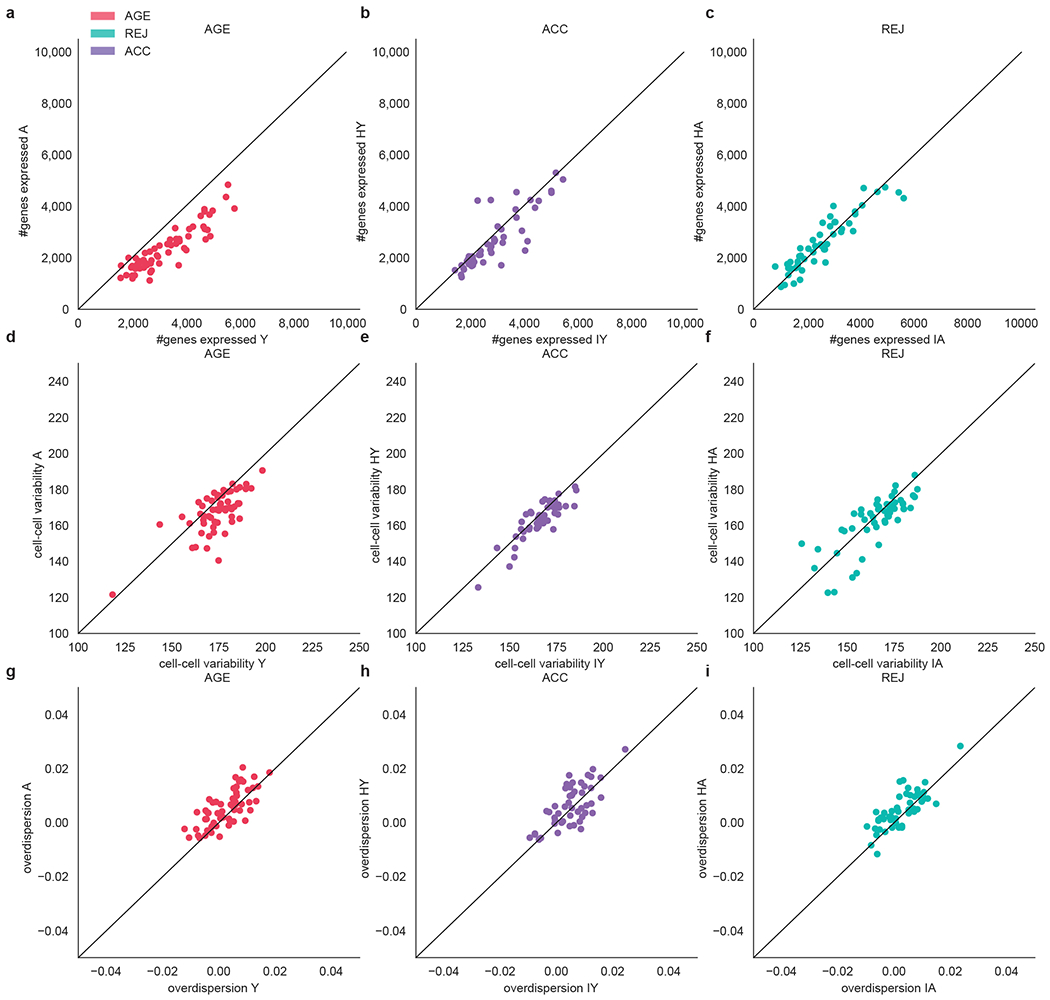
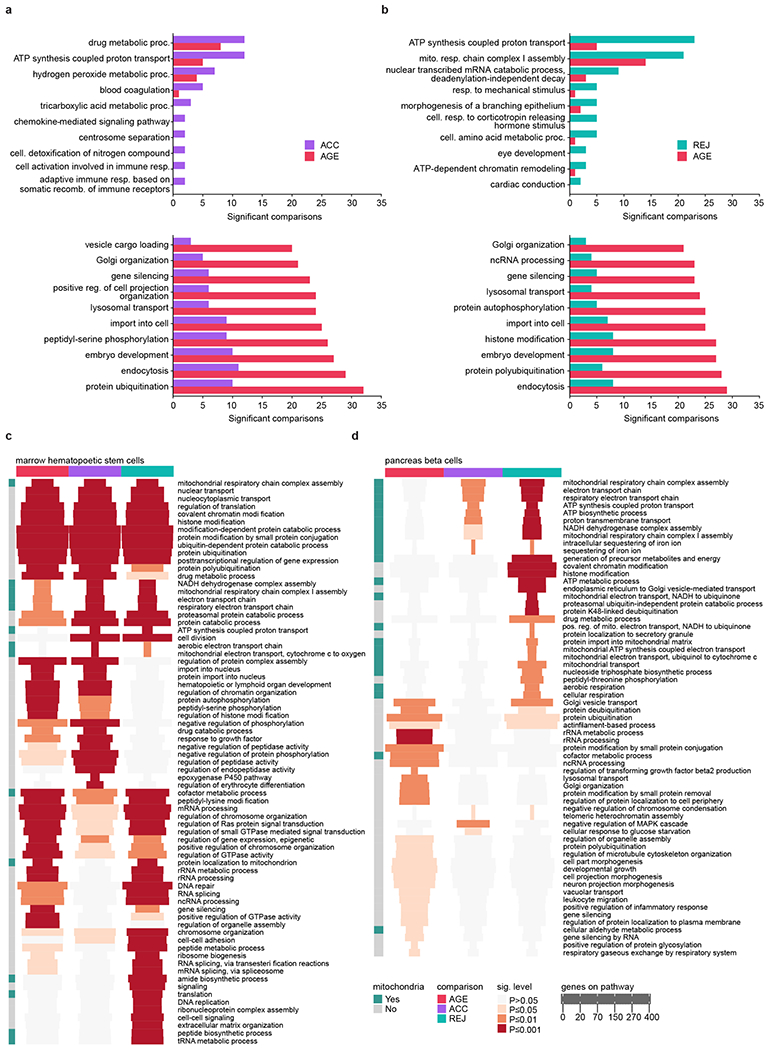
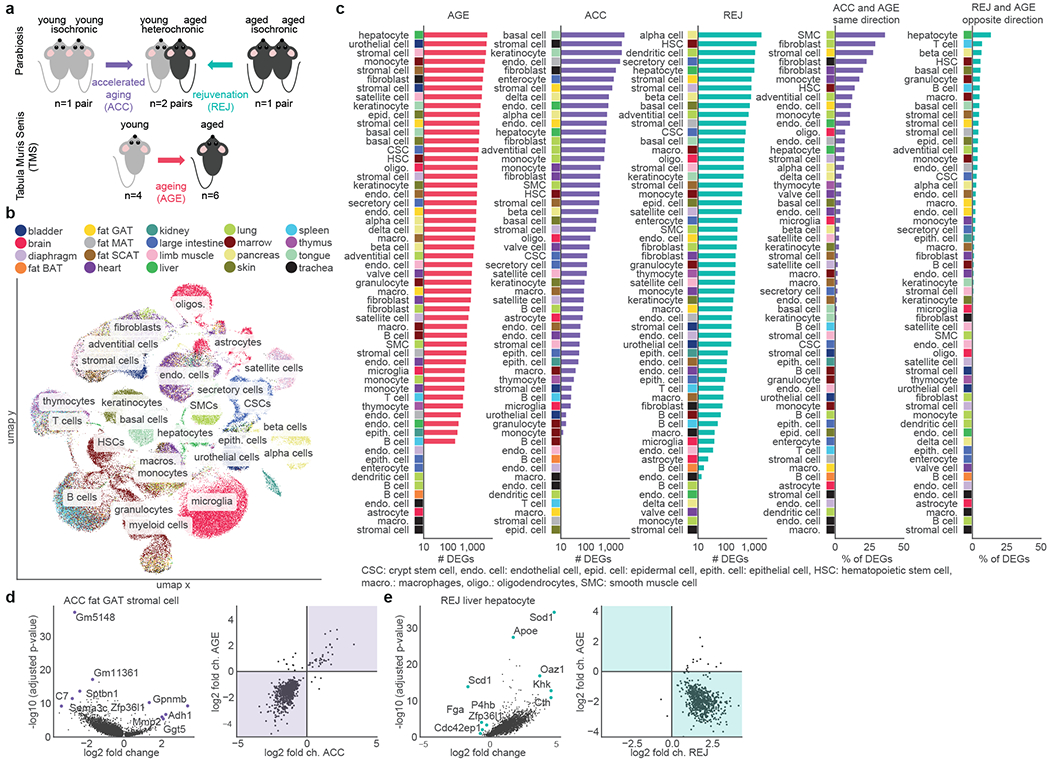
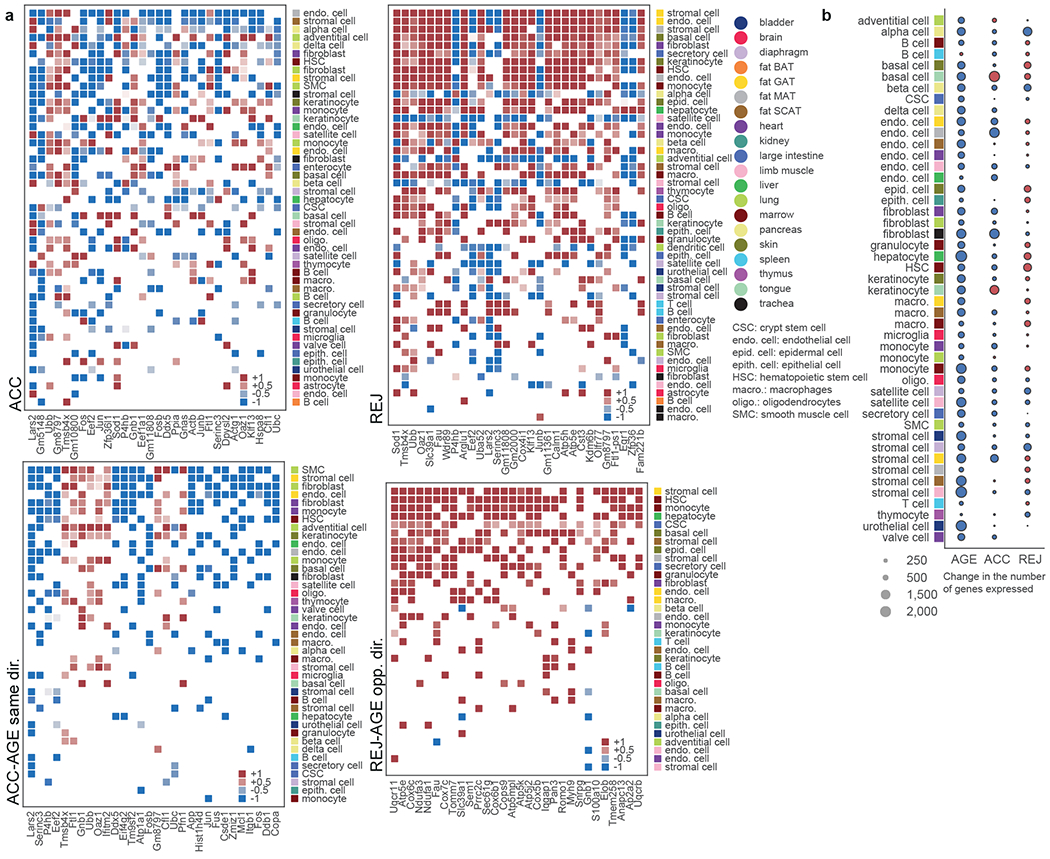
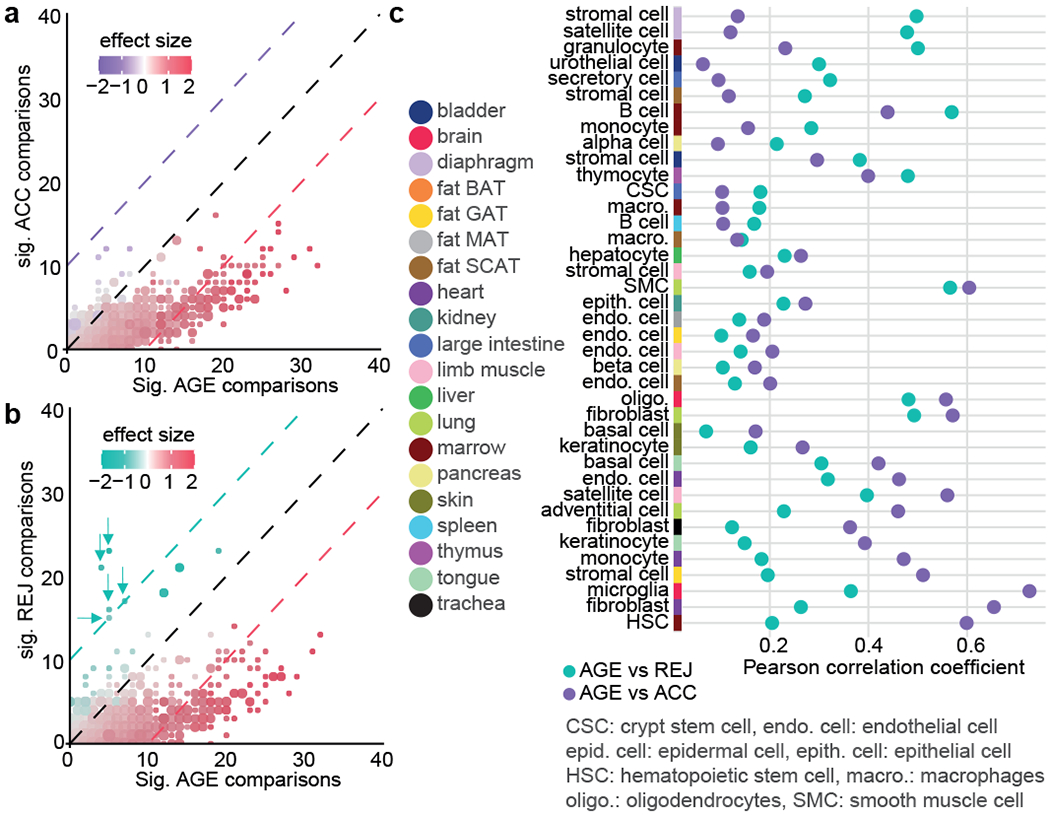
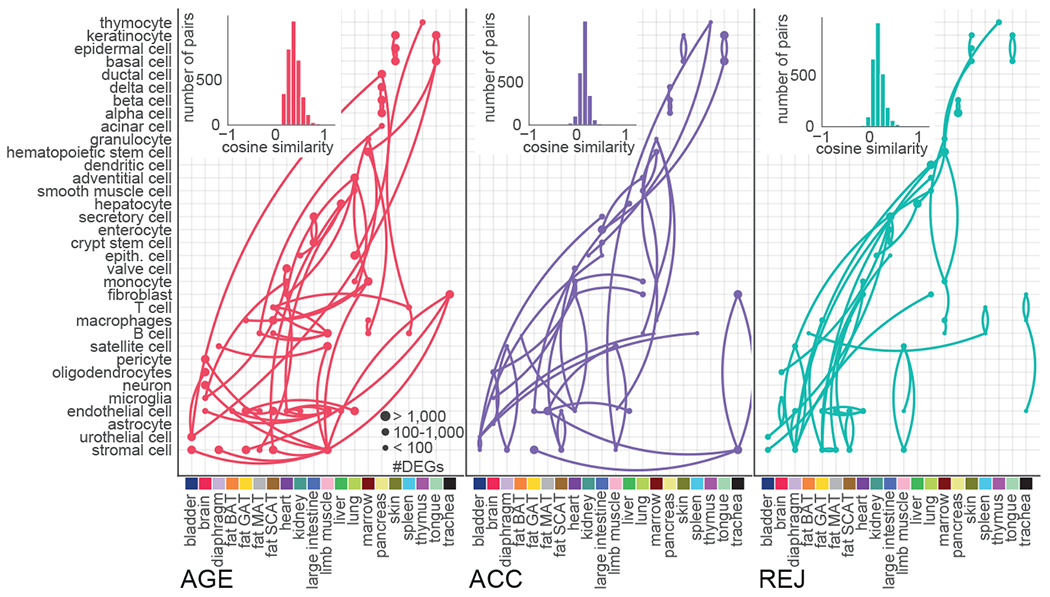
The ability to slow or reverse biological ageing would have major implications for mitigating disease risk and maintaining vitality1. Although an increasing number of interventions show promise for rejuvenation2, their effectiveness on disparate cell types across the body and the molecular pathways susceptible to rejuvenation remain largely unexplored. Here we performed single-cell RNA sequencing on 20 organs to reveal cell-type-specific responses to young and aged blood in heterochronic parabiosis. Adipose mesenchymal stromal cells, haematopoietic stem cells and hepatocytes are among those cell types that are especially responsive. On the pathway level, young blood invokes new gene sets in addition to reversing established ageing patterns, with the global rescue of genes encoding electron transport chain subunits pinpointing a prominent role of mitochondrial function in parabiosis-mediated rejuvenation. We observed an almost universal loss of gene expression with age that is largely mimicked by parabiosis: aged blood reduces global gene expression, and young blood restores it in select cell types. Together, these data lay the groundwork for a systemic understanding of the interplay between blood-borne factors and cellular integrity.
© 2022. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Competing interests
The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper.
Figures

References
Methods References
-
- Picelli S et al. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc 9, 171–181 (2014). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
