

Sampling alternative conformational states of transporters and receptors with AlphaFold2
- PMID: 35238773
- PMCID: PMC9023059
- DOI: 10.7554/eLife.75751
Sampling alternative conformational states of transporters and receptors with AlphaFold2
Abstract
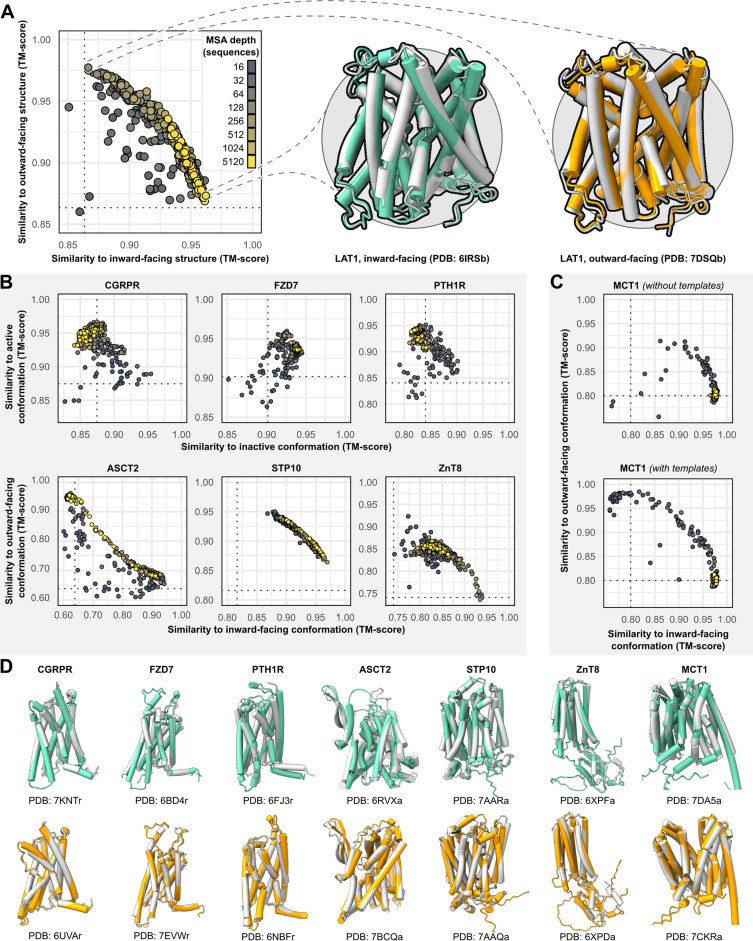
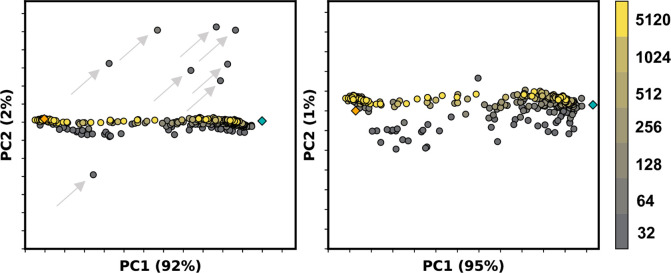
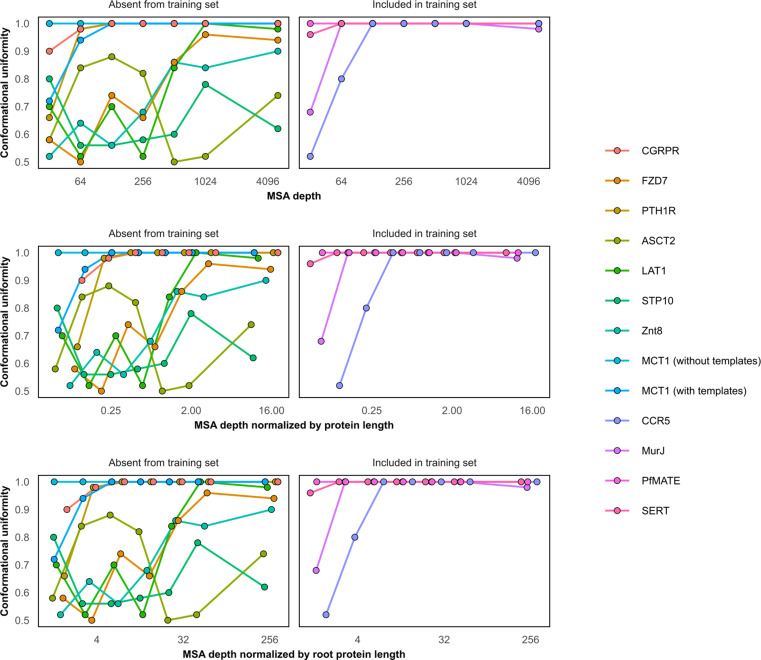
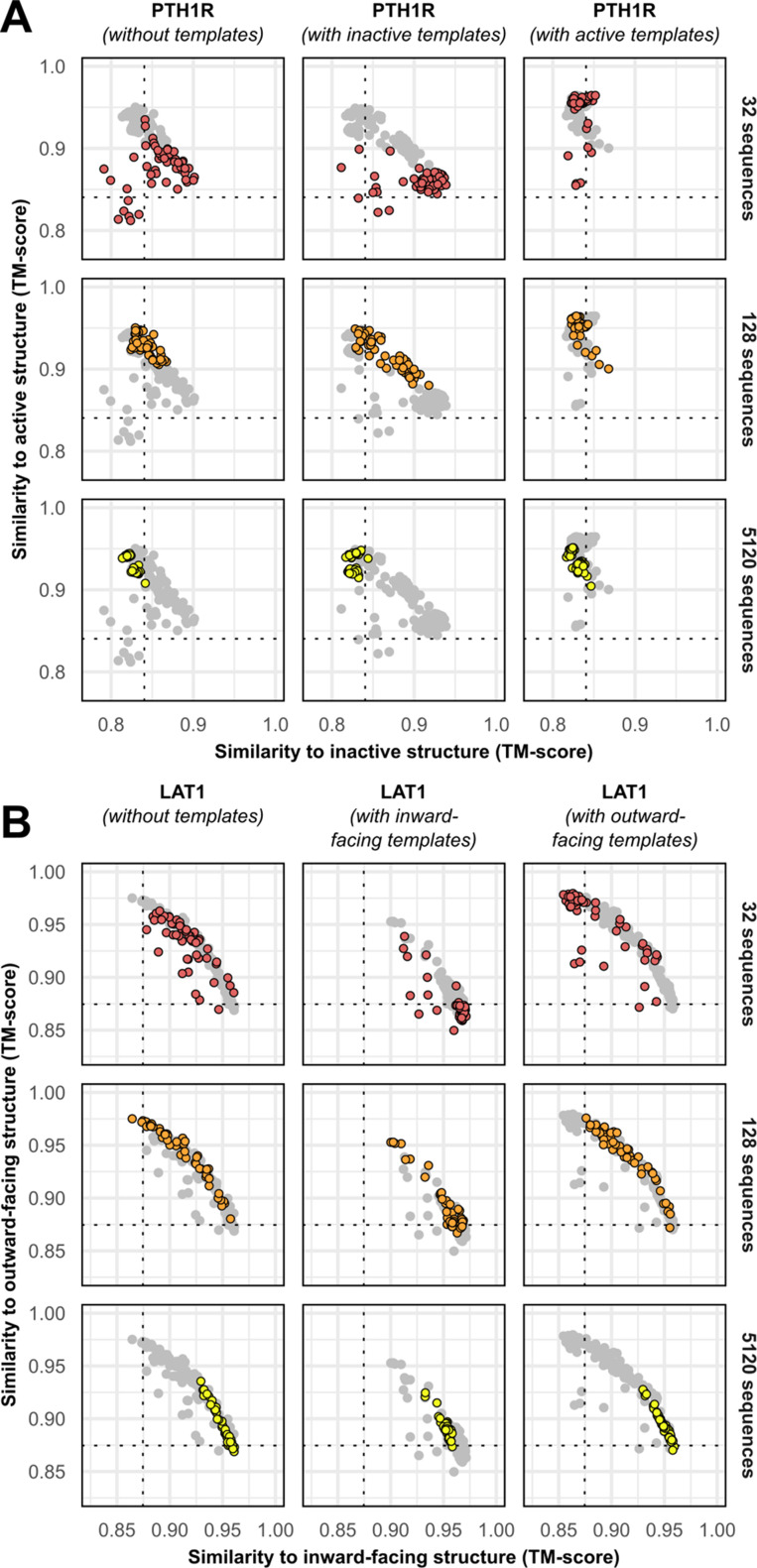
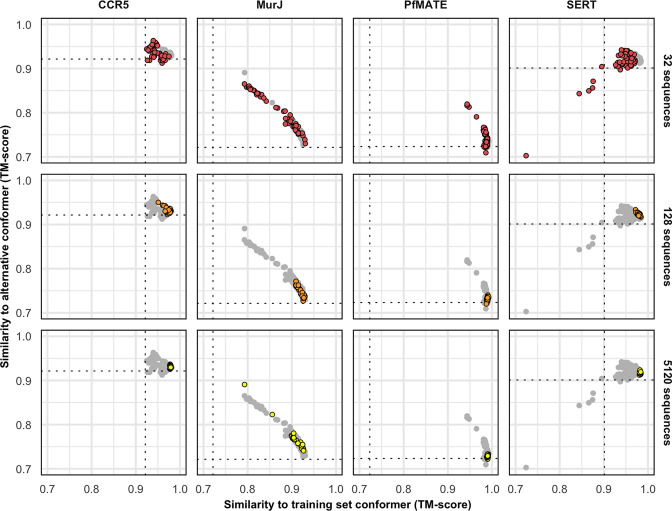
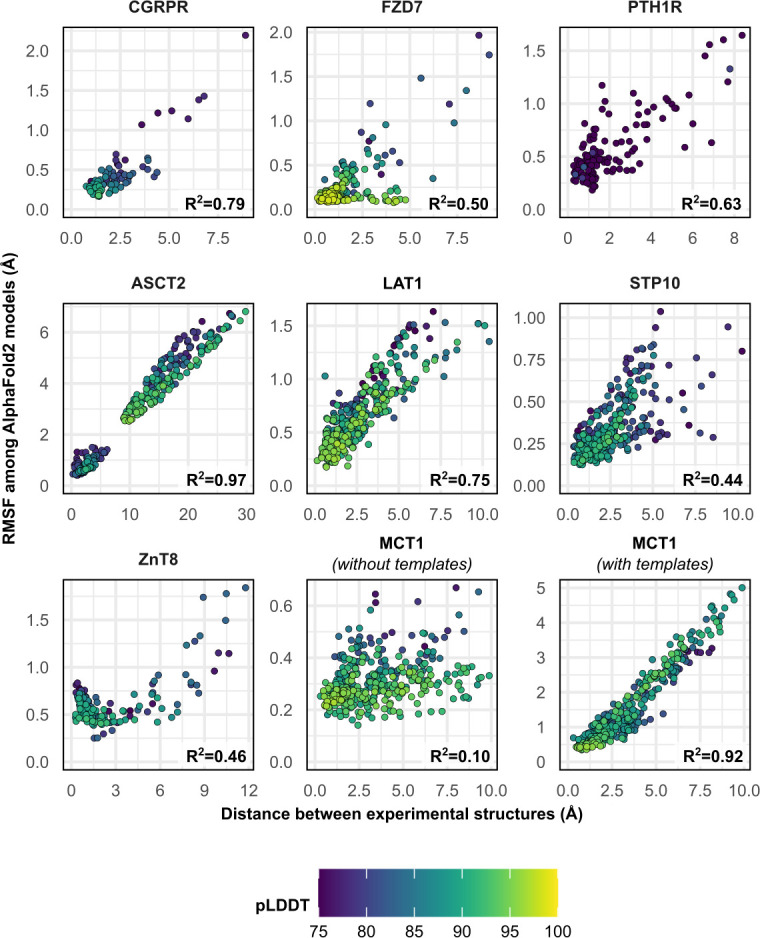
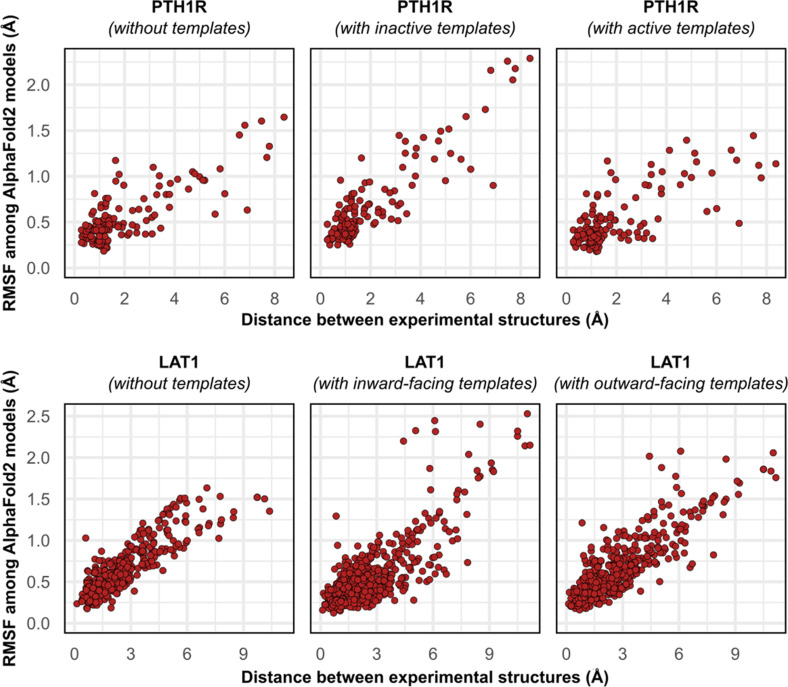
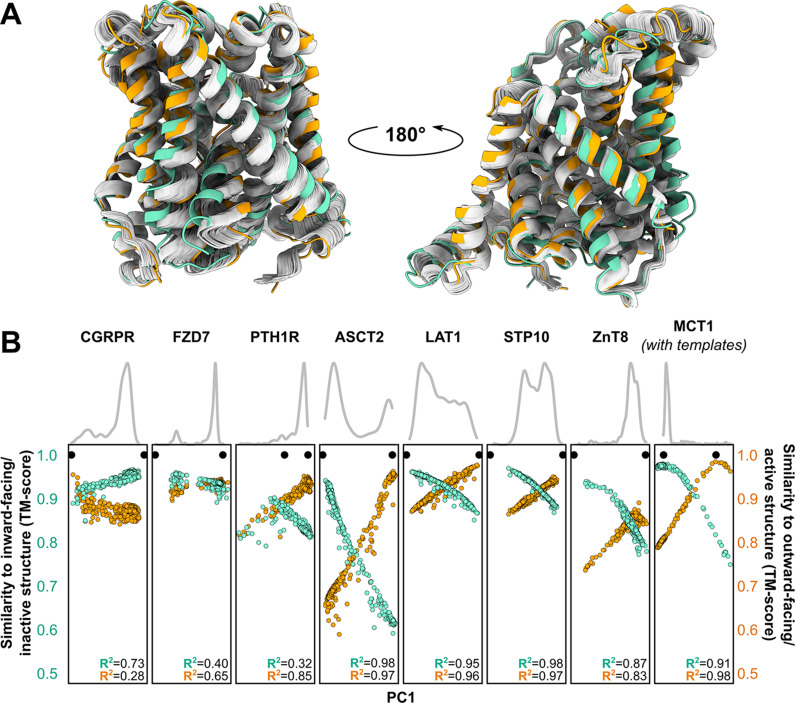
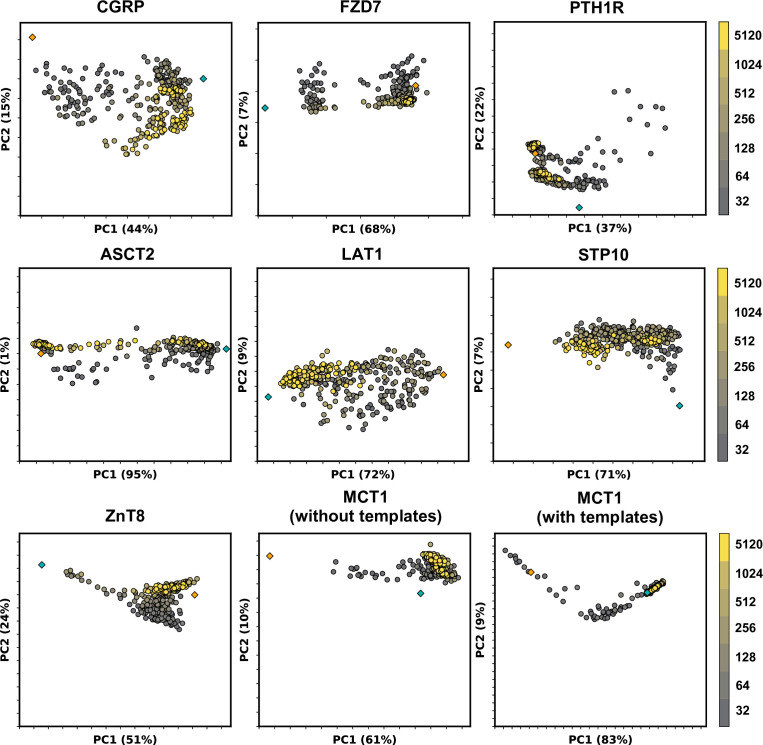
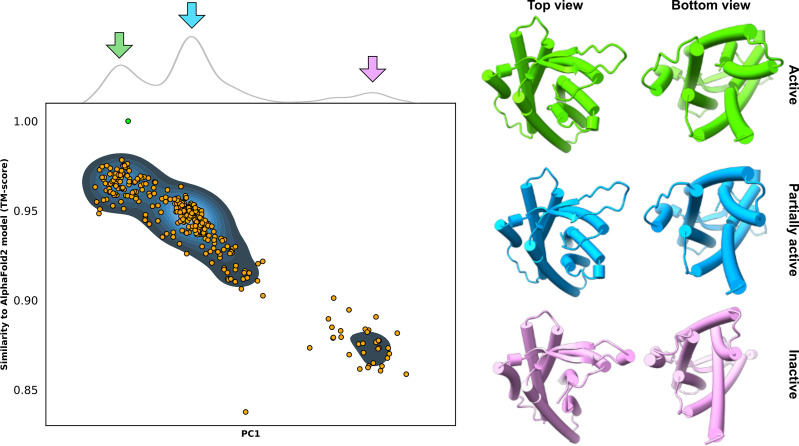
Equilibrium fluctuations and triggered conformational changes often underlie the functional cycles of membrane proteins. For example, transporters mediate the passage of molecules across cell membranes by alternating between inward- and outward-facing states, while receptors undergo intracellular structural rearrangements that initiate signaling cascades. Although the conformational plasticity of these proteins has historically posed a challenge for traditional de novo protein structure prediction pipelines, the recent success of AlphaFold2 (AF2) in CASP14 culminated in the modeling of a transporter in multiple conformations to high accuracy. Given that AF2 was designed to predict static structures of proteins, it remains unclear if this result represents an underexplored capability to accurately predict multiple conformations and/or structural heterogeneity. Here, we present an approach to drive AF2 to sample alternative conformations of topologically diverse transporters and G-protein-coupled receptors that are absent from the AF2 training set. Whereas models of most proteins generated using the default AF2 pipeline are conformationally homogeneous and nearly identical to one another, reducing the depth of the input multiple sequence alignments by stochastic subsampling led to the generation of accurate models in multiple conformations. In our benchmark, these conformations spanned the range between two experimental structures of interest, with models at the extremes of these conformational distributions observed to be among the most accurate (average template modeling score of 0.94). These results suggest a straightforward approach to identifying native-like alternative states, while also highlighting the need for the next generation of deep learning algorithms to be designed to predict ensembles of biophysically relevant states.
Keywords: G-protein-coupled receptors; conformational dynamics; molecular biophysics; none; protein structure prediction; structural biology; transporters.
© 2022, del Alamo et al.
Conflict of interest statement
Dd, DS, HM, JM No competing interests declared
Figures
Comment in
- doi: 10.7554/eLife.78549
References
-
- Akdel M, Pires DEV, Porta Pardo E, Jänes J, Zalevsky AO, Mészáros B, Bryant P, Good LL, Laskowski RA, Pozzati G, Shenoy A, Zhu W, Kundrotas P, Ruiz Serra V, Rodrigues CHM, Dunham AS, Burke D, Borkakoti N, Velankar S, Frost A, Lindorff-Larsen K, Valencia A, Ovchinnikov S, Durairaj J, Ascher DB, Thornton JM, Davey NE, Stein A, Elofsson A, Croll TI, Beltrao P. A structural biology community assessment of AlphaFold 2 applications. bioRxiv. 2021 doi: 10.1101/2021.09.26.461876. - DOI - PMC - PubMed
-
- Campbell E, Kaltenbach M, Correy GJ, Carr PD, Porebski BT, Livingstone EK, Afriat-Jurnou L, Buckle AM, Weik M, Hollfelder F, Tokuriki N, Jackson CJ. The role of protein dynamics in the evolution of new enzyme function. Nature Chemical Biology. 2016;12:944–950. doi: 10.1038/nchembio.2175. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
