

Transcriptional neighborhoods regulate transcript isoform lengths and expression levels
- PMID: 35239377
- PMCID: PMC7613581
- DOI: 10.1126/science.abg0162
Transcriptional neighborhoods regulate transcript isoform lengths and expression levels
Abstract
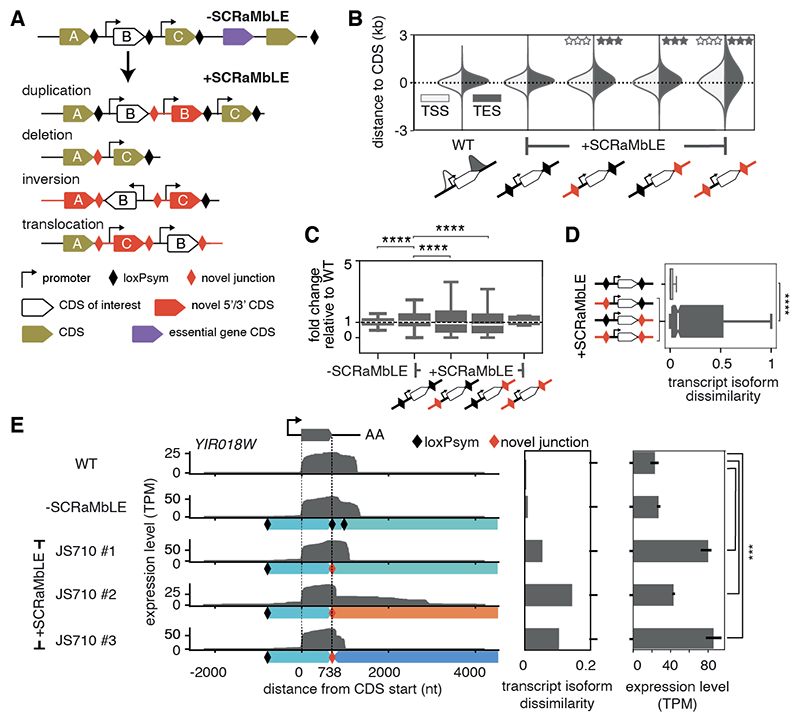
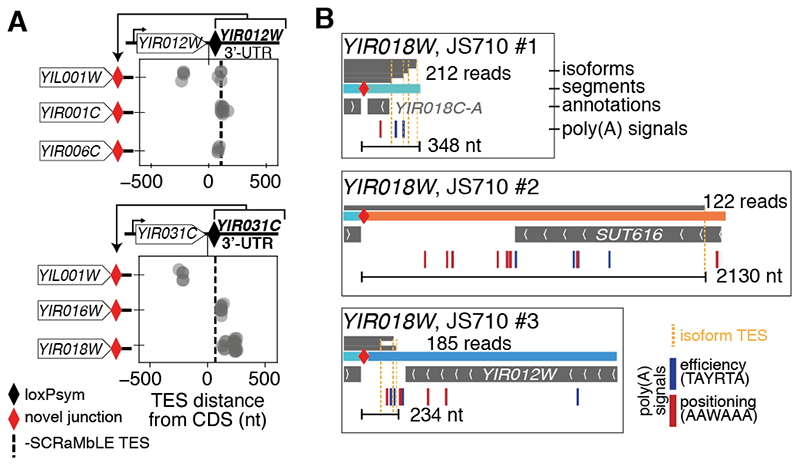
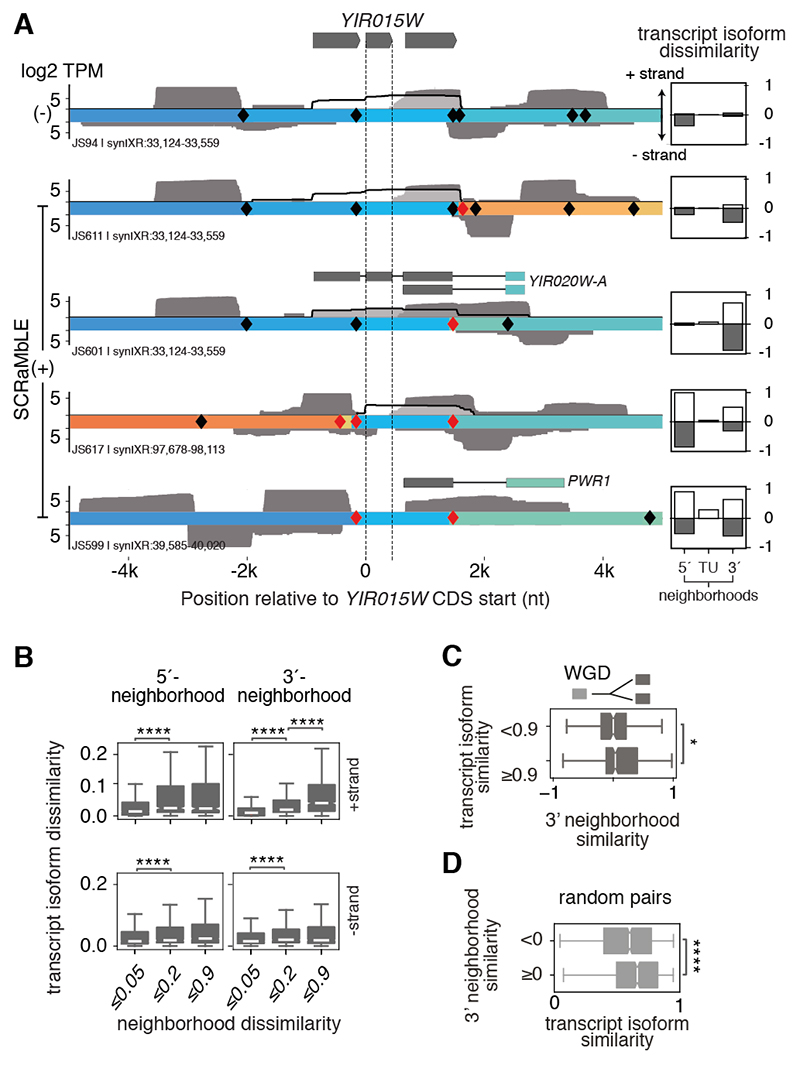
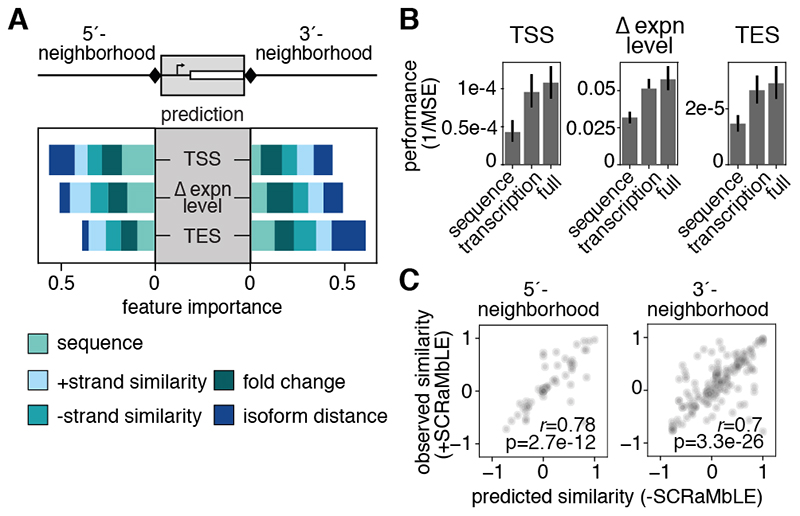
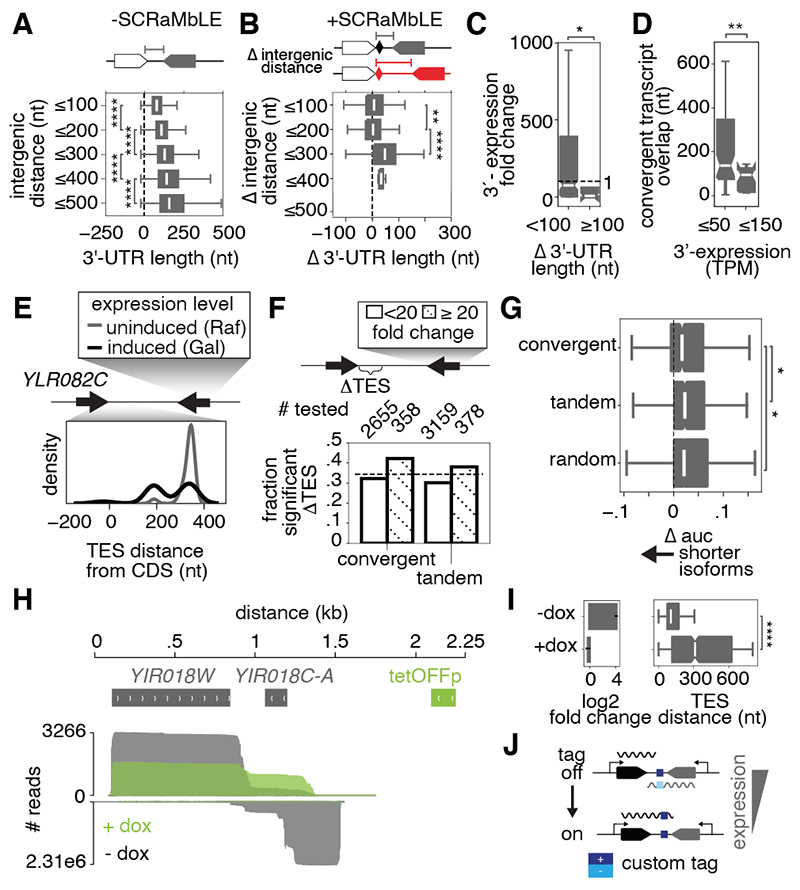
Sequence features of genes and their flanking regulatory regions are determinants of RNA transcript isoform expression and have been used as context-independent plug-and-play modules in synthetic biology. However, genetic context-including the adjacent transcriptional environment-also influences transcript isoform expression levels and boundaries. We used synthetic yeast strains with stochastically repositioned genes to systematically disentangle the effects of sequence and context. Profiling 120 million full-length transcript molecules across 612 genomic perturbations, we observed sequence-independent alterations to gene expression levels and transcript isoform boundaries that were influenced by neighboring transcription. We identified features of transcriptional context that could predict these alterations and used these features to engineer a synthetic circuit where transcript length was controlled by neighboring transcription. This demonstrates how positional context can be leveraged in synthetic genome engineering.
Conflict of interest statement
Figures
Comment in
-
Neighbourly modulation of transcript isoforms.Nat Rev Genet. 2022 May;23(5):264. doi: 10.1038/s41576-022-00476-7. Nat Rev Genet. 2022. PMID: 35314808 No abstract available.
References
-
- Danino YM, Even D, Ideses D, Juven-Gershon T. The core promoter: At the heart of gene expression. Biochim Biophys Acta. 2015;1849:1116–1131. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
