

Emerging Methods and Applications to Decrypt Allostery in Proteins and Nucleic Acids
- PMID: 35240127
- PMCID: PMC9398933
- DOI: 10.1016/j.jmb.2022.167518
Emerging Methods and Applications to Decrypt Allostery in Proteins and Nucleic Acids
Abstract
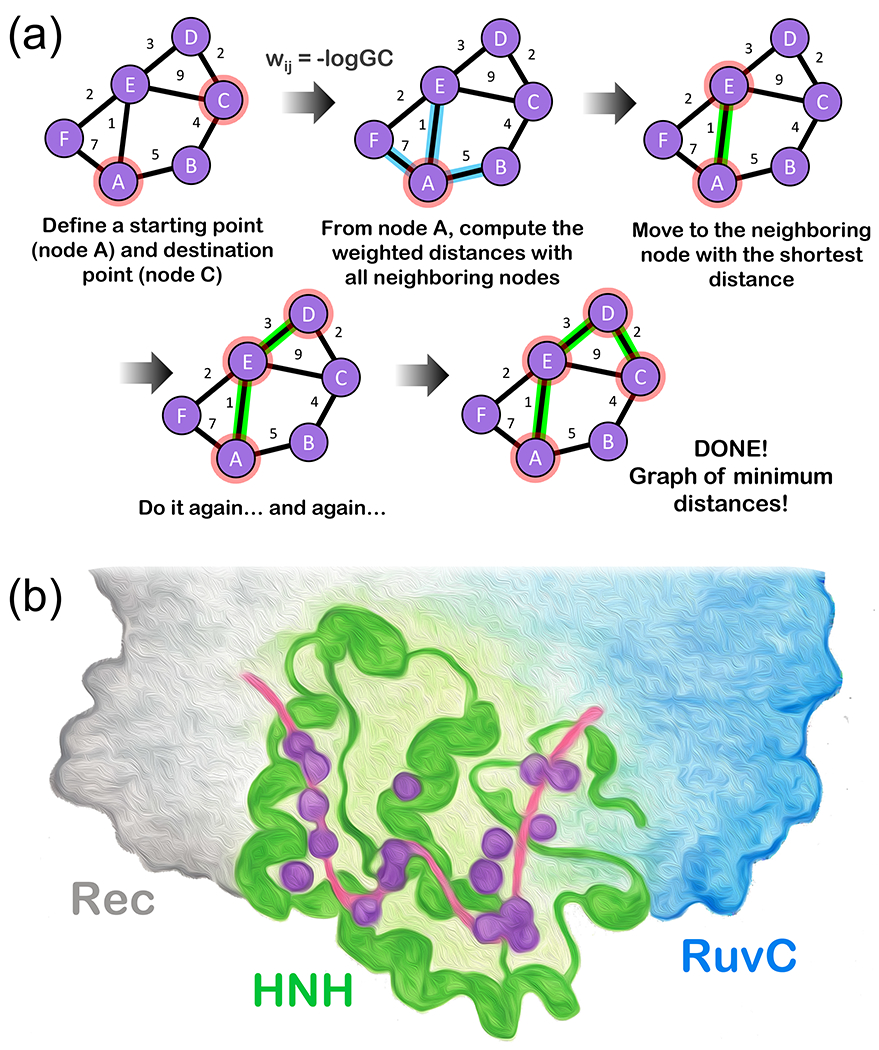
Many large protein-nucleic acid complexes exhibit allosteric regulation. In these systems, the propagation of the allosteric signaling is strongly coupled to conformational dynamics and catalytic function, challenging state-of-the-art analytical methods. Here, we review established and innovative approaches used to elucidate allosteric mechanisms in these complexes. Specifically, we report network models derived from graph theory and centrality analyses in combination with molecular dynamics (MD) simulations, introducing novel schemes that implement the synergistic use of graph theory with enhanced simulations methods and ab-initio MD. Accelerated MD simulations are used to construct "enhanced network models", describing the allosteric response over long timescales and capturing the relation between allostery and conformational changes. "Ab-initio network models" combine graph theory with ab-initio MD and quantum mechanics/molecular mechanics (QM/MM) simulations to describe the allosteric regulation of catalysis by following the step-by-step dynamics of biochemical reactions. This approach characterizes how the allosteric regulation changes from reactants to products and how it affects the transition state, revealing a tense-to-relaxed allosteric regulation along the chemical step. Allosteric models and applications are showcased for three paradigmatic examples of allostery in protein-nucleic acid complexes: (i) the nucleosome core particle, (ii) the CRISPR-Cas9 genome editing system and (iii) the spliceosome. These methods and applications create innovative protocols to determine allosteric mechanisms in protein-nucleic acid complexes that show tremendous promise for medicine and bioengineering.
Keywords: CRISPR-Cas9; graph theory; molecular dynamics; nucleosome core particle; spliceosome.
Copyright © 2022 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Competing Interests The authors declare no competing financial interest.
Figures







Similar articles
-
Dynamics and mechanisms of CRISPR-Cas9 through the lens of computational methods.Curr Opin Struct Biol. 2022 Aug;75:102400. doi: 10.1016/j.sbi.2022.102400. Epub 2022 Jun 8. Curr Opin Struct Biol. 2022. PMID: 35689914 Free PMC article. Review.
-
Establishing the allosteric mechanism in CRISPR-Cas9.Wiley Interdiscip Rev Comput Mol Sci. 2021 May-Jun;11(3):e1503. doi: 10.1002/wcms.1503. Epub 2020 Oct 26. Wiley Interdiscip Rev Comput Mol Sci. 2021. PMID: 34322166 Free PMC article.
-
Understanding the mechanistic basis of non-coding RNA through molecular dynamics simulations.J Struct Biol. 2019 Jun 1;206(3):267-279. doi: 10.1016/j.jsb.2019.03.004. Epub 2019 Mar 15. J Struct Biol. 2019. PMID: 30880083 Free PMC article. Review.
-
Markov models for the elucidation of allosteric regulation.Philos Trans R Soc Lond B Biol Sci. 2018 Jun 19;373(1749):20170178. doi: 10.1098/rstb.2017.0178. Philos Trans R Soc Lond B Biol Sci. 2018. PMID: 29735732 Free PMC article. Review.
-
CRISPR-Cas9 conformational activation as elucidated from enhanced molecular simulations.Proc Natl Acad Sci U S A. 2017 Jul 11;114(28):7260-7265. doi: 10.1073/pnas.1707645114. Epub 2017 Jun 26. Proc Natl Acad Sci U S A. 2017. PMID: 28652374 Free PMC article.
Cited by
-
Graph Attention Neural Networks Reveal TnsC Filament Assembly in a CRISPR-Associated Transposon.bioRxiv [Preprint]. 2025 Jun 17:2025.06.17.659969. doi: 10.1101/2025.06.17.659969. bioRxiv. 2025. PMID: 40666904 Free PMC article. Preprint.
-
New design strategies for ultra-specific CRISPR-Cas13a-based RNA detection with single-nucleotide mismatch sensitivity.Nucleic Acids Res. 2024 Jan 25;52(2):921-939. doi: 10.1093/nar/gkad1132. Nucleic Acids Res. 2024. PMID: 38033324 Free PMC article.
-
Unveiling the RNA-mediated allosteric activation discloses functional hotspots in CRISPR-Cas13a.Nucleic Acids Res. 2024 Jan 25;52(2):906-920. doi: 10.1093/nar/gkad1127. Nucleic Acids Res. 2024. PMID: 38033317 Free PMC article.
-
Machine learning and protein allostery.Trends Biochem Sci. 2023 Apr;48(4):375-390. doi: 10.1016/j.tibs.2022.12.001. Epub 2022 Dec 21. Trends Biochem Sci. 2023. PMID: 36564251 Free PMC article. Review.
-
Evolutionary rewiring of the dynamic network underpinning allosteric epistasis in NS1 of the influenza A virus.Proc Natl Acad Sci U S A. 2025 Feb 25;122(8):e2410813122. doi: 10.1073/pnas.2410813122. Epub 2025 Feb 20. Proc Natl Acad Sci U S A. 2025. PMID: 39977319 Free PMC article.
