

Clinical and genetic characteristics of two patients with tyrosinemia type 1 in Slovenia - A novel fumarylacetoacetate hydrolase (FAH) intronic disease-causing variant
- PMID: 35242570
- PMCID: PMC8856938
- DOI: 10.1016/j.ymgmr.2021.100836
Clinical and genetic characteristics of two patients with tyrosinemia type 1 in Slovenia - A novel fumarylacetoacetate hydrolase (FAH) intronic disease-causing variant
Abstract
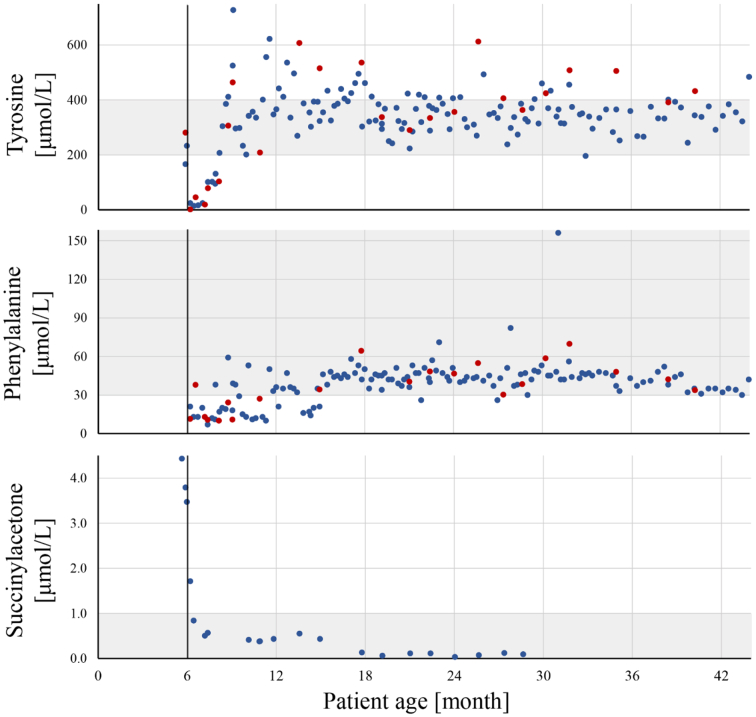
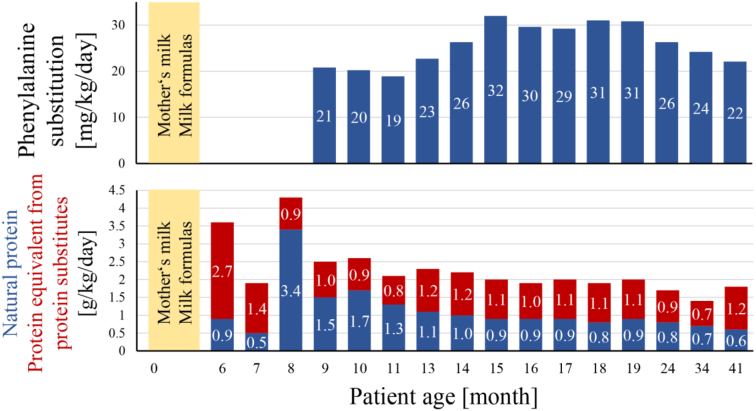
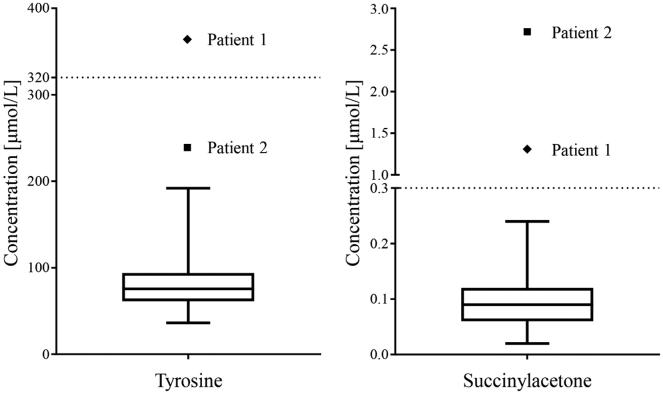
Tyrosinemia type 1 (HT1) is an inborn error of tyrosine catabolism that leads to severe liver, kidney, and neurological dysfunction. Newborn screening (NBS) can enable a timely diagnosis and early initiation of treatment. We presented the follow up of the only two Slovenian patients diagnosed with HT1. Metabolic control was monitored by measuring tyrosine, phenylalanine and succinylacetone from dried blood spots (DBSs). Retrograde screening of HT1 was performed from DBSs taken at birth using tandem mass spectrometry. First patient was diagnosed at the age of 6 months in the asymptomatic phase due to an abnormal liver echogenicity, the other presented at 2.5 months with an acute liver failure and needed a liver transplantation. The first was a compound heterozygote for a novel FAH intronic variant c.607-21A>G and c.192G>T whereas the second was homozygous for c.192G>T. At the non-transplanted patient, 66% of tyrosine and 79% of phenylalanine measurements were in strict reference ranges of 200-400 μmol/L and >30 μmol/L, respectively, which resulted in a favorable cognitive outcome at 3.6 years. On retrograde screening, both patients had elevated SA levels; on the other hand, tyrosine was elevated only at one. We showed that non-coding regions should be analyzed when clinical and biochemical markers are characteristic of HT1. DBSs represent a convenient sample type for frequent amino acid monitoring. Retrograde diagnosis of HT1 was possible after more than three years of birth with SA as a primary marker, complemented by tyrosine.
Keywords: AFP, alpha-fetoprotein; ALP, alkaline phosphatase; ALT, alanine transaminase; AST, aspartate transaminase; DBS, dried blood spot; Dried blood spot; FAH, fumarylacetoacetate hydrolase; Fumarylacetoacetate hydrolase; GGT, gamma glutamyl transferase; HT1, tyrosinemia type 1; INR, international normalized ratio; Intronic variant; MS/MS, tandem mass spectrometry; NBS, newborn screening; NTBC, nitisinone; Nitisinone; PTT, partial thromboplastin time; RF, reference range; SA, succinylacetone; Succinylacetone; Tyrosinemia.
© 2021 The Authors.
Conflict of interest statement
Authors declare no conflicts of interest.
Figures
Similar articles
-
Mildly elevated succinylacetone and normal liver function in compound heterozygotes with pathogenic and pseudodeficient FAH alleles.Mol Genet Metab Rep. 2017 Dec 27;14:55-58. doi: 10.1016/j.ymgmr.2017.12.002. eCollection 2018 Mar. Mol Genet Metab Rep. 2017. PMID: 29326876 Free PMC article.
-
Diagnosis and the importance of early treatment of tyrosinemia type 1: A case report.Clin Mass Spectrom. 2019 Feb 2;12:1-6. doi: 10.1016/j.clinms.2019.01.005. eCollection 2019 Apr. Clin Mass Spectrom. 2019. PMID: 34841073 Free PMC article.
-
A Lithuanian Case of Tyrosinemia Type 1 with a Literature Review: A Rare Cause of Acute Liver Failure in Childhood.Medicina (Kaunas). 2024 Jan 11;60(1):135. doi: 10.3390/medicina60010135. Medicina (Kaunas). 2024. PMID: 38256395 Free PMC article.
-
Biochemical and Clinical Aspects of Hereditary Tyrosinemia Type 1.Adv Exp Med Biol. 2017;959:9-21. doi: 10.1007/978-3-319-55780-9_2. Adv Exp Med Biol. 2017. PMID: 28755181 Review.
-
Newborn Screening for Hereditary Tyrosinemia Type I in Québec: Update.Adv Exp Med Biol. 2017;959:139-146. doi: 10.1007/978-3-319-55780-9_13. Adv Exp Med Biol. 2017. PMID: 28755192 Review.
Cited by
-
Identification and functional characterization of a novel homozygous intronic variant in the fumarylacetoacetate hydrolase gene in a Chinese patient with tyrosinemia type 1.BMC Med Genomics. 2022 Dec 3;15(1):251. doi: 10.1186/s12920-022-01406-6. BMC Med Genomics. 2022. PMID: 36463171 Free PMC article.
References
-
- van Spronsen F.J., Thomasse Y., Smit G.P.A., Leonard J.V., Clayton P.T., Fidler V., et al. Hereditary tyrosinemia type I: a new clinical classification with difference in prognosis on dietary treatment. Hepatology. 1994;20(5):1187–1191. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
