

Circuit topology analysis of cellular genome reveals signature motifs, conformational heterogeneity, and scaling
- PMID: 35243229
- PMCID: PMC8861635
- DOI: 10.1016/j.isci.2022.103866
Circuit topology analysis of cellular genome reveals signature motifs, conformational heterogeneity, and scaling
Abstract
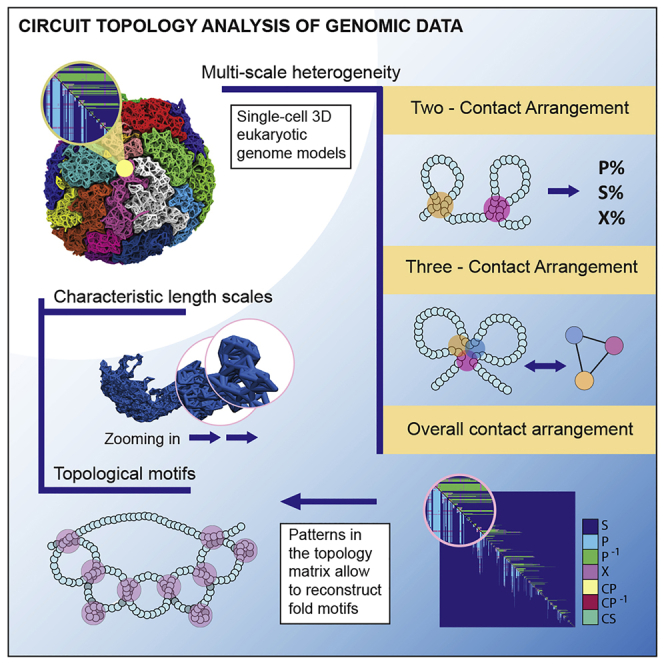
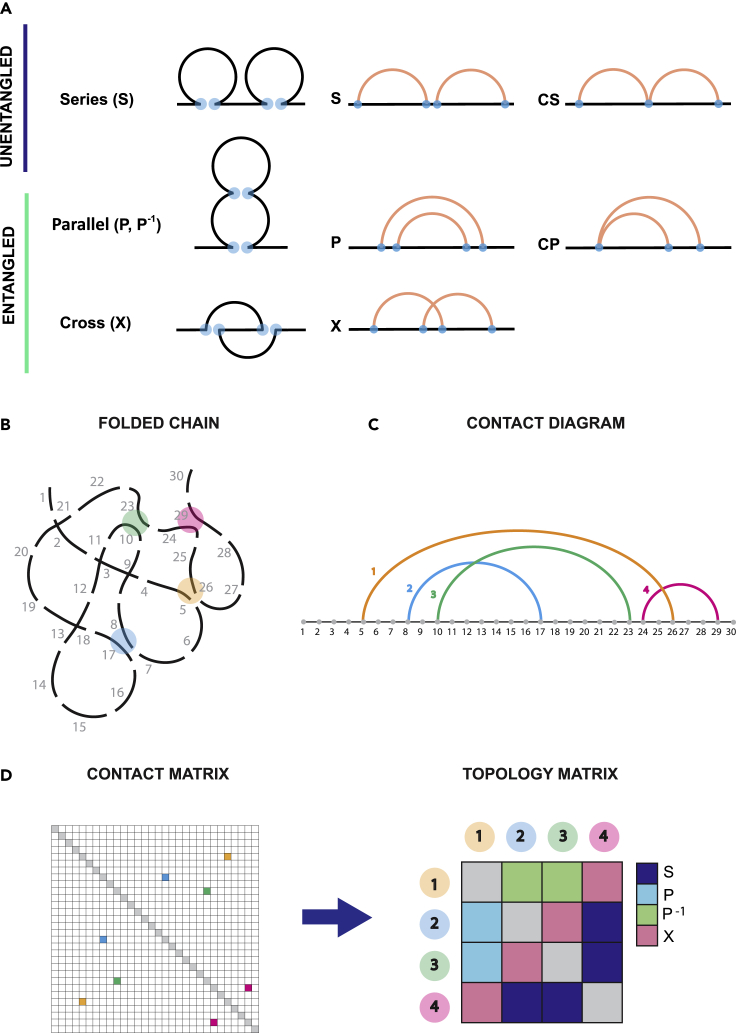
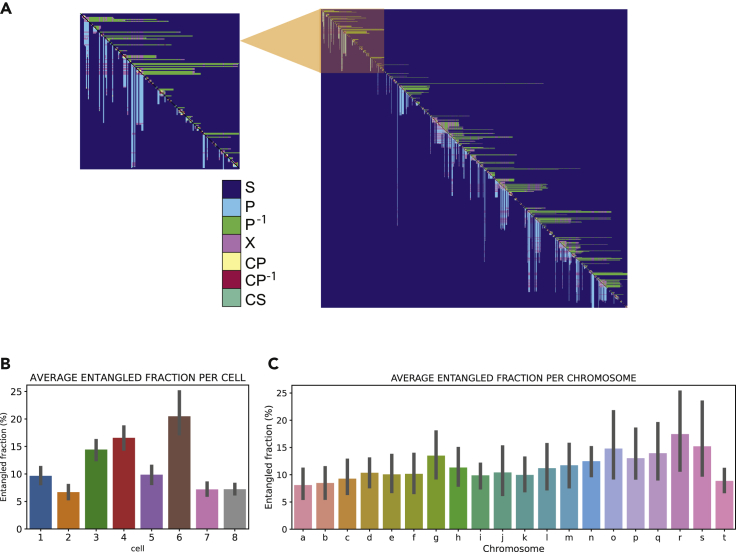
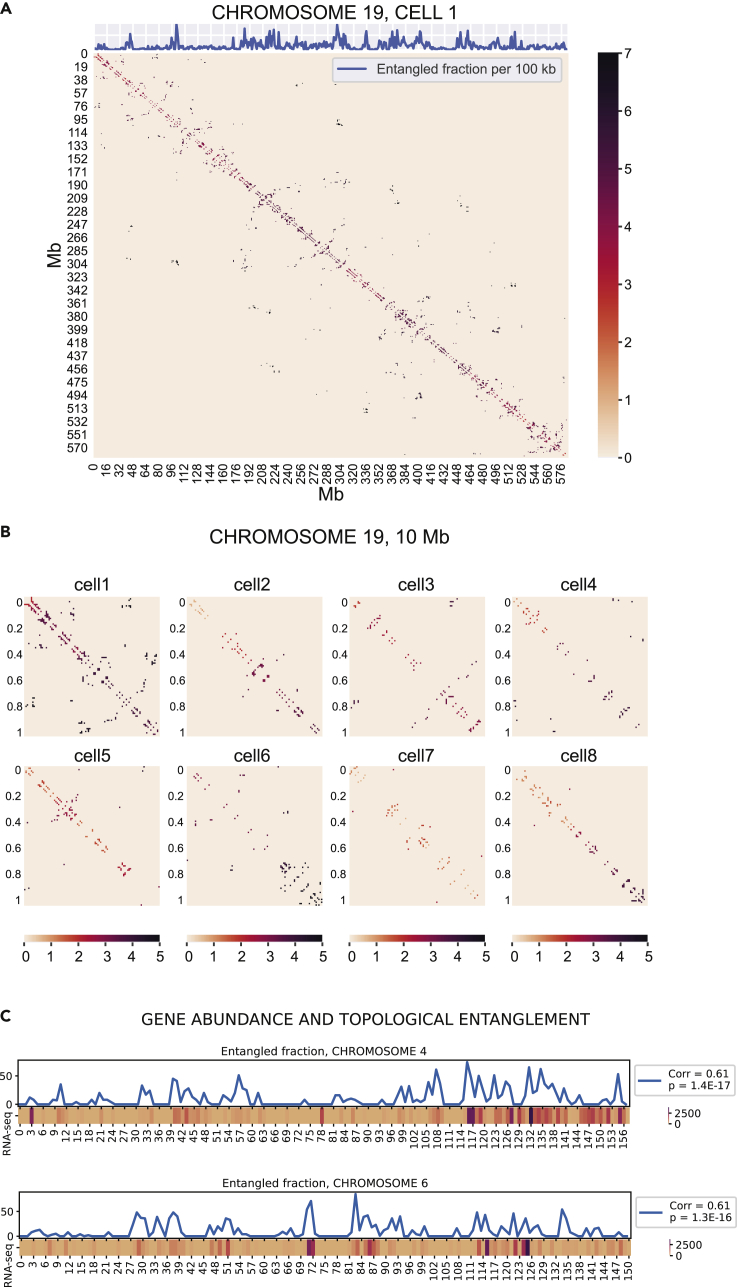
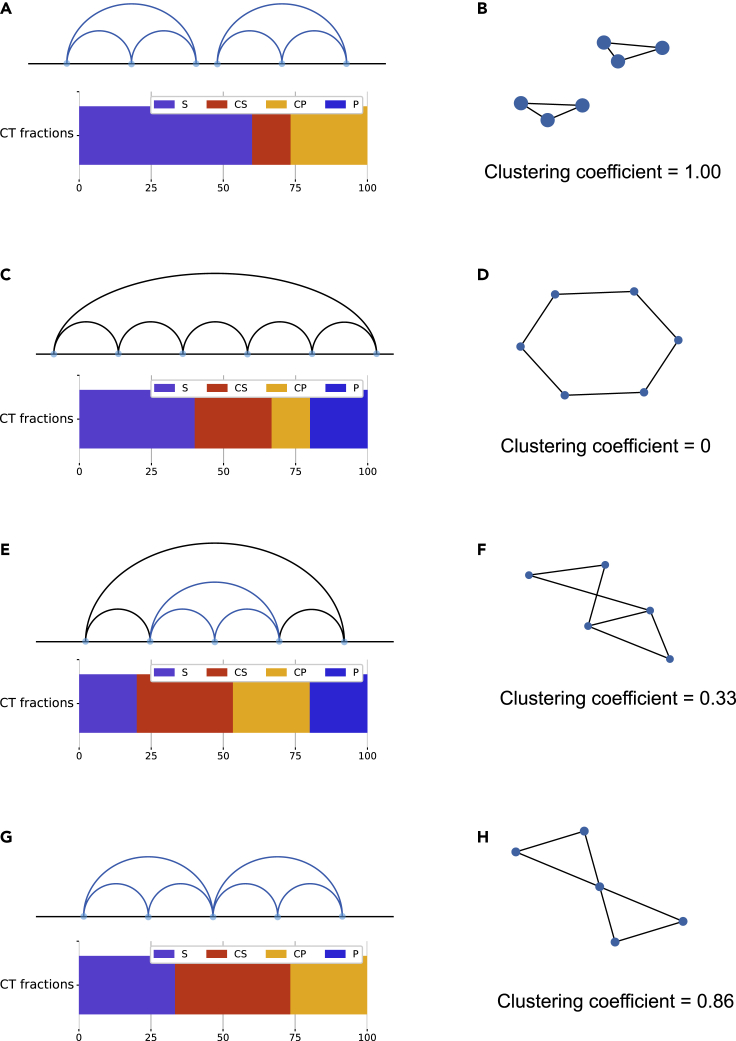
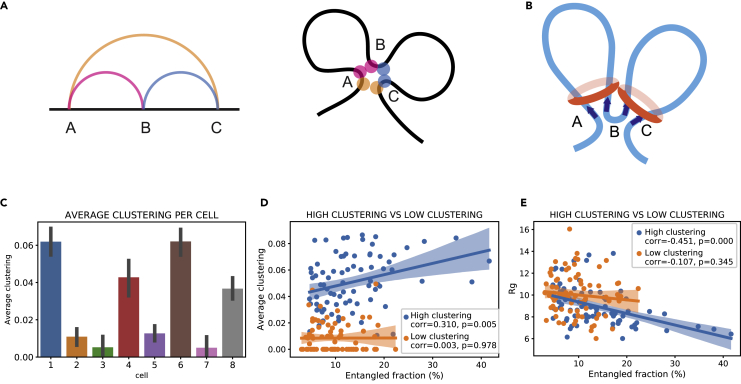
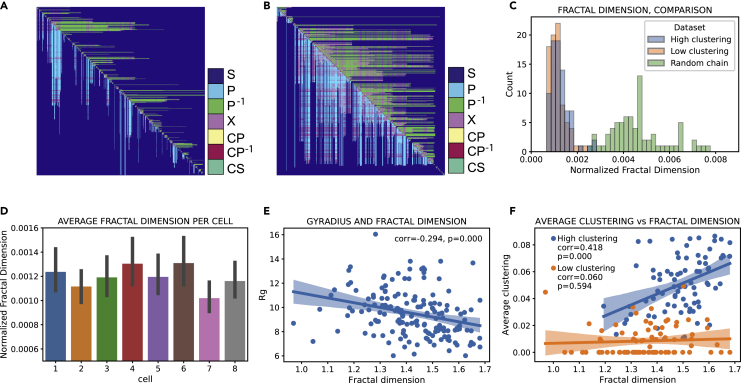
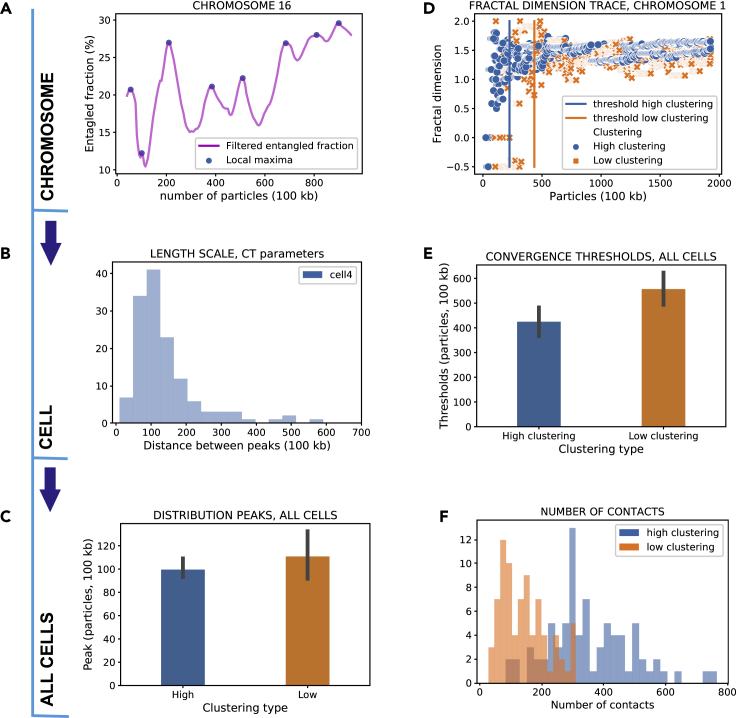
Reciprocal regulation of genome topology and function is a fundamental and enduring puzzle in biology. The wealth of data provided by Hi-C libraries offers the opportunity to unravel this relationship. However, there is a need for a comprehensive theoretical framework in order to extract topological information for genome characterization and comparison. Here, we develop a toolbox for topological analysis based on Circuit Topology, allowing for the quantification of inter- and intracellular genomic heterogeneity, at various levels of fold complexity: pairwise contact arrangement, higher-order contact arrangement, and topological fractal dimension. Single-cell Hi-C data were analyzed and characterized based on topological content, revealing not only a strong multiscale heterogeneity but also highly conserved features such as a characteristic topological length scale and topological signature motifs in the genome. We propose that these motifs inform on the topological state of the nucleus and indicate the presence of active loop extrusion.
Keywords: Genomics; In silico biology; Mathematical biosciences.
© 2022 The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
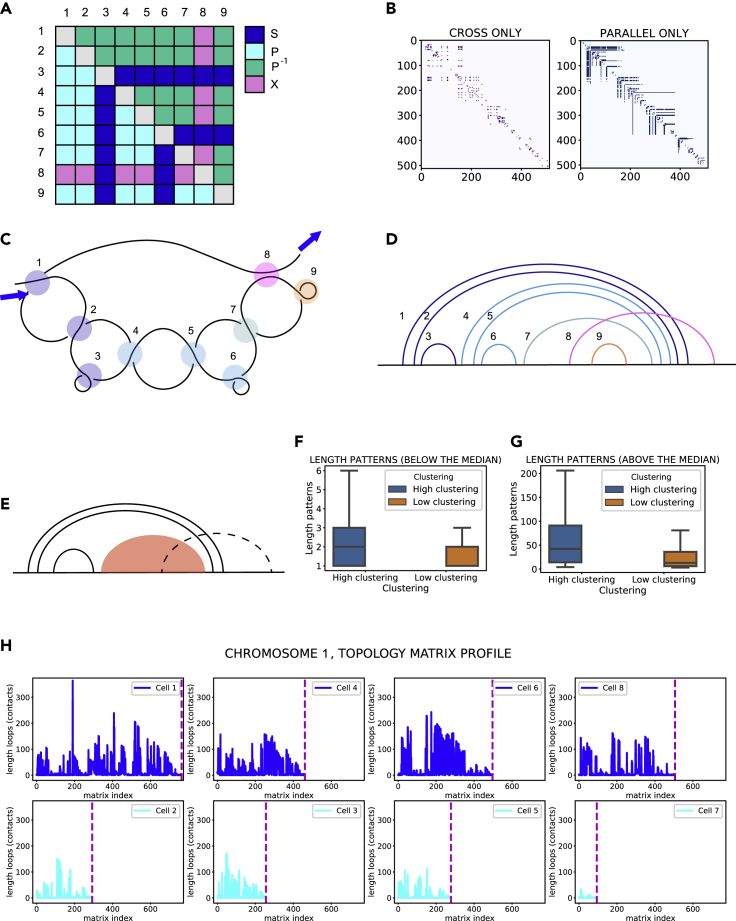
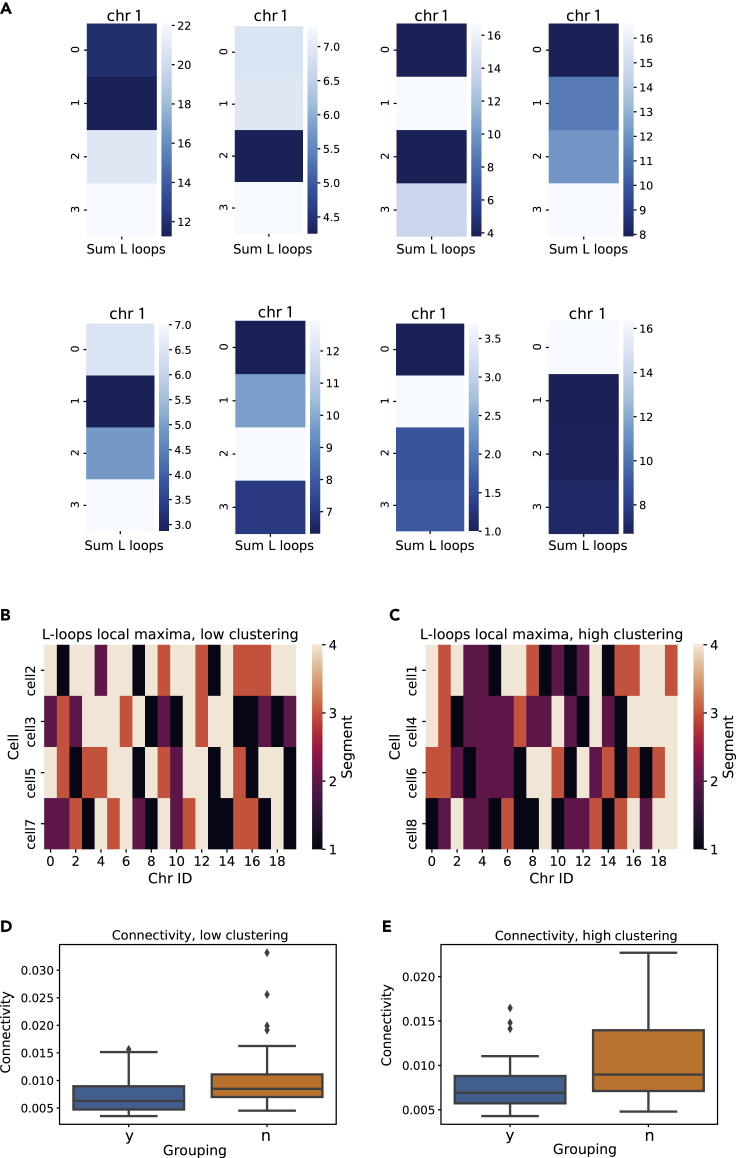
Similar articles
-
Circuit Topology Analysis of Single-Cell HiC Data.Methods Mol Biol. 2024;2819:27-38. doi: 10.1007/978-1-0716-3930-6_2. Methods Mol Biol. 2024. PMID: 39028500
-
Current theoretical models fail to predict the topological complexity of the human genome.Front Mol Biosci. 2015 Aug 21;2:48. doi: 10.3389/fmolb.2015.00048. eCollection 2015. Front Mol Biosci. 2015. PMID: 26347874 Free PMC article.
-
Aggregation and structural phase transitions of semiflexible polymer bundles: A braided circuit topology approach.iScience. 2024 Jan 26;27(3):108995. doi: 10.1016/j.isci.2024.108995. eCollection 2024 Mar 15. iScience. 2024. PMID: 38361617 Free PMC article.
-
Topological portraits of multiscale coordination dynamics.J Neurosci Methods. 2020 Jun 1;339:108672. doi: 10.1016/j.jneumeth.2020.108672. Epub 2020 Mar 6. J Neurosci Methods. 2020. PMID: 32151601 Free PMC article. Review.
-
Multiscale geometric and topological analyses for characterizing and predicting immune responses from single cell data.Trends Immunol. 2023 Jul;44(7):551-563. doi: 10.1016/j.it.2023.05.003. Epub 2023 Jun 9. Trends Immunol. 2023. PMID: 37301677 Review.
Cited by
-
ProteinCT: An implementation of the protein circuit topology framework.MethodsX. 2022 Sep 16;9:101861. doi: 10.1016/j.mex.2022.101861. eCollection 2022. MethodsX. 2022. PMID: 36187158 Free PMC article.
-
Circuit Topology Analysis of Single-Cell HiC Data.Methods Mol Biol. 2024;2819:27-38. doi: 10.1007/978-1-0716-3930-6_2. Methods Mol Biol. 2024. PMID: 39028500
References
-
- Carrière M., Rabadán R. In: Topological Data Analysis. Abel Symposia. Baas N., Carlsson G., Quick G., Szymik M., editors. Springer; 2020. Topological data analysis of single-cell hi-C contact maps; pp. 147–162. - DOI
LinkOut - more resources
Full Text Sources
