

Comprehensive genetic diagnosis of tandem repeat expansion disorders with programmable targeted nanopore sequencing
- PMID: 35245110
- PMCID: PMC8896783
- DOI: 10.1126/sciadv.abm5386
Comprehensive genetic diagnosis of tandem repeat expansion disorders with programmable targeted nanopore sequencing
Abstract
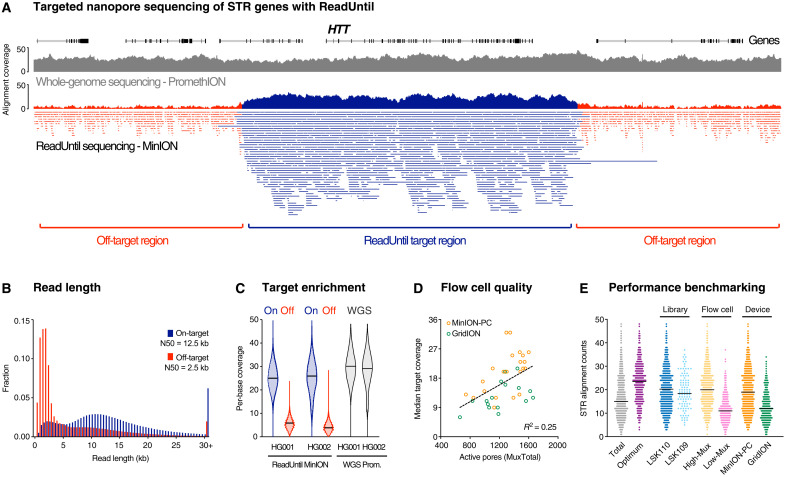
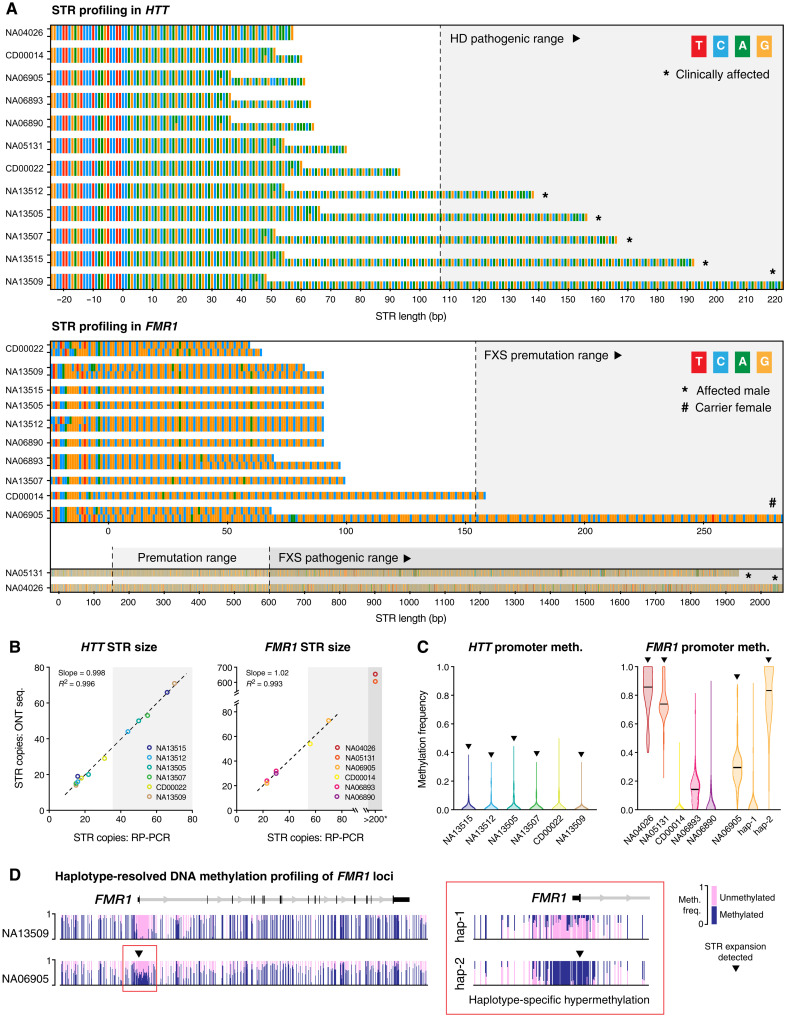
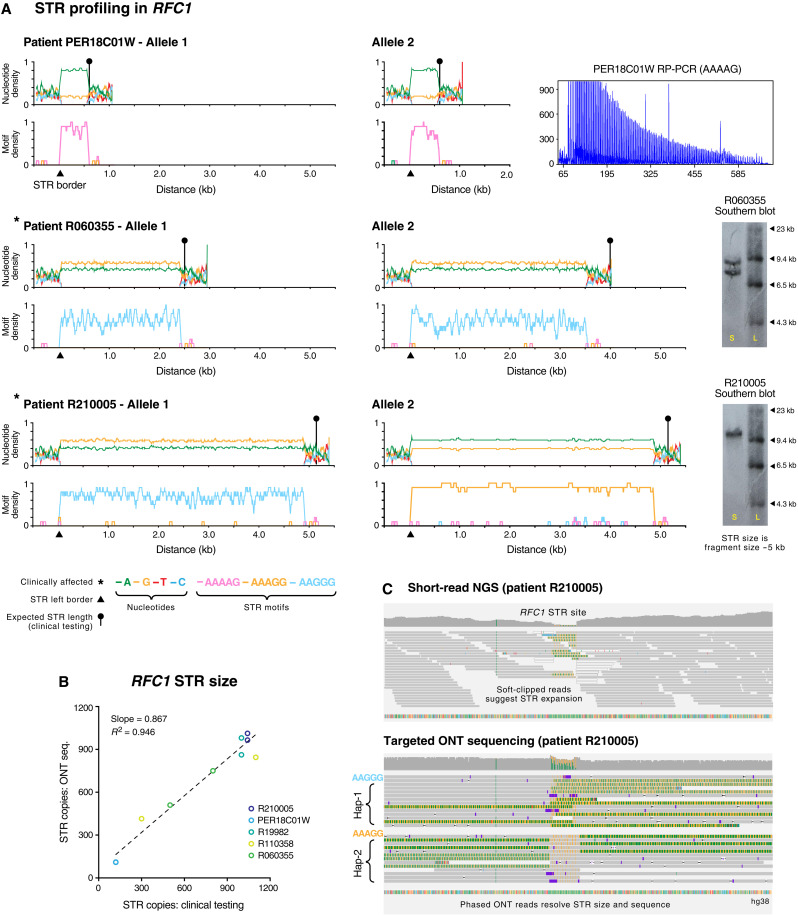
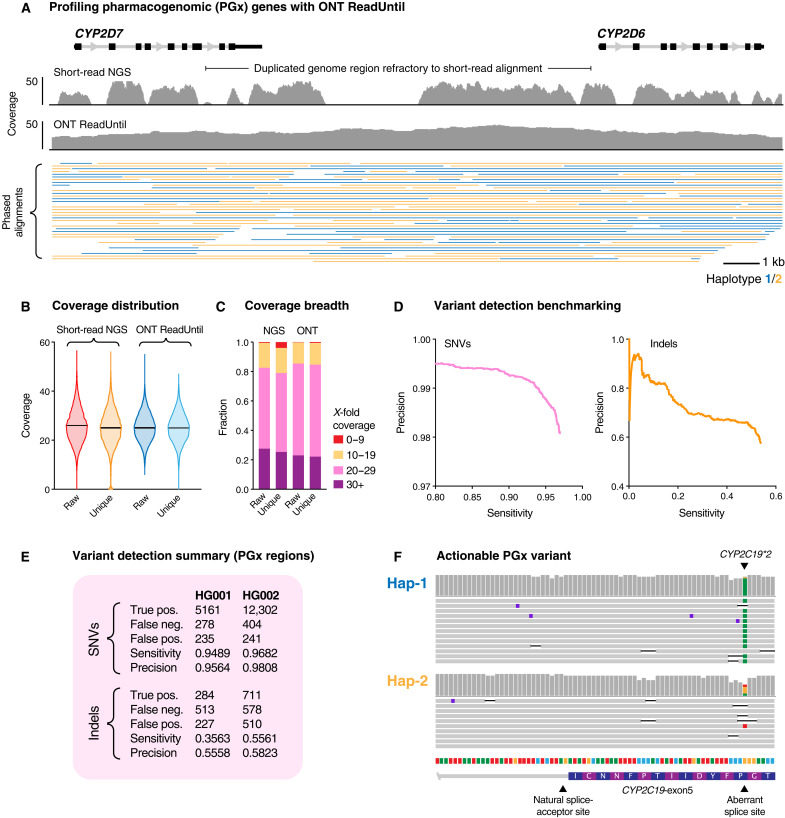
More than 50 neurological and neuromuscular diseases are caused by short tandem repeat (STR) expansions, with 37 different genes implicated to date. We describe the use of programmable targeted long-read sequencing with Oxford Nanopore's ReadUntil function for parallel genotyping of all known neuropathogenic STRs in a single assay. Our approach enables accurate, haplotype-resolved assembly and DNA methylation profiling of STR sites, from a list of predetermined candidates. This correctly diagnoses all individuals in a small cohort (n = 37) including patients with various neurogenetic diseases (n = 25). Targeted long-read sequencing solves large and complex STR expansions that confound established molecular tests and short-read sequencing and identifies noncanonical STR motif conformations and internal sequence interruptions. We observe a diversity of STR alleles of known and unknown pathogenicity, suggesting that long-read sequencing will redefine the genetic landscape of repeat disorders. Last, we show how the inclusion of pharmacogenomic genes as secondary ReadUntil targets can further inform patient care.
Figures

Similar articles
-
Nanopore Long-Read Sequencing as a First-Tier Diagnostic Test to Detect Repeat Expansions in Neurological Disorders.Int J Mol Sci. 2025 Mar 21;26(7):2850. doi: 10.3390/ijms26072850. Int J Mol Sci. 2025. PMID: 40243408 Free PMC article.
-
An update on the neurological short tandem repeat expansion disorders and the emergence of long-read sequencing diagnostics.Acta Neuropathol Commun. 2021 May 25;9(1):98. doi: 10.1186/s40478-021-01201-x. Acta Neuropathol Commun. 2021. PMID: 34034831 Free PMC article. Review.
-
Profiling of Short-Tandem-Repeat Disease Alleles in 12,632 Human Whole Genomes.Am J Hum Genet. 2017 Nov 2;101(5):700-715. doi: 10.1016/j.ajhg.2017.09.013. Am J Hum Genet. 2017. PMID: 29100084 Free PMC article.
-
NanoMnT: an STR analysis tool for Oxford Nanopore sequencing data driven by a comprehensive analysis of error profile in STR regions.Gigascience. 2025 Jan 6;14:giaf013. doi: 10.1093/gigascience/giaf013. Gigascience. 2025. PMID: 40094553 Free PMC article.
-
Advances in forensic genetics: Exploring the potential of long read sequencing.Forensic Sci Int Genet. 2025 Jan;74:103156. doi: 10.1016/j.fsigen.2024.103156. Epub 2024 Oct 10. Forensic Sci Int Genet. 2025. PMID: 39427416 Review.
Cited by
-
STRling: a k-mer counting approach that detects short tandem repeat expansions at known and novel loci.Genome Biol. 2022 Dec 14;23(1):257. doi: 10.1186/s13059-022-02826-4. Genome Biol. 2022. PMID: 36517892 Free PMC article.
-
Genewise detection of variants in MEFV gene using nanopore sequencing.Front Genet. 2024 Nov 29;15:1493295. doi: 10.3389/fgene.2024.1493295. eCollection 2024. Front Genet. 2024. PMID: 39678383 Free PMC article.
-
Origin and evolution of pentanucleotide repeat expansions at the familial cortical myoclonic tremor with epilepsy type1 - SAMD12 locus.Eur J Hum Genet. 2025 Mar;33(2):252-257. doi: 10.1038/s41431-024-01586-y. Epub 2024 Mar 12. Eur J Hum Genet. 2025. PMID: 38467733
-
Expanding horizons of tandem repeats in biology and medicine: Why 'genomic dark matter' matters.Emerg Top Life Sci. 2023 Dec 13;7(3):239-47. doi: 10.1042/ETLS20230075. Online ahead of print. Emerg Top Life Sci. 2023. PMID: 38088823 Free PMC article.
-
Enhancing Molecular Testing for Effective Delivery of Actionable Gene Diagnostics.Bioengineering (Basel). 2022 Dec 1;9(12):745. doi: 10.3390/bioengineering9120745. Bioengineering (Basel). 2022. PMID: 36550951 Free PMC article. Review.
References
-
- Ellegren H., Microsatellites: Simple sequences with complex evolution. Nat. Rev. Genet. 5, 435–445 (2004). - PubMed
-
- Hantash F. M., Goos D. M., Crossley B., Anderson B., Zhang K., Sun W., Strom C. M., FMR1 premutation carrier frequency in patients undergoing routine population-based carrier screening: Insights into the prevalence of fragile X syndrome, fragile X-associated tremor/ataxia syndrome, and fragile X-associated primary ovarian insufficiency in the United States. Genet. Med. 13, 39–45 (2011). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
