

Bi-allelic mutation of CTNNB1 causes a severe form of syndromic microphthalmia, persistent foetal vasculature and vitreoretinal dysplasia
- PMID: 35246174
- PMCID: PMC8896279
- DOI: 10.1186/s13023-022-02239-3
Bi-allelic mutation of CTNNB1 causes a severe form of syndromic microphthalmia, persistent foetal vasculature and vitreoretinal dysplasia
Abstract
Background: Inherited vitreoretinopathies arise as a consequence of congenital retinal vascularisation abnormalities. They represent a phenotypically and genetically heterogeneous group of disorders that can have a major impact on vision. Several genes encoding proteins and effectors of the canonical Wnt/β-catenin pathway have been associated and precise diagnosis, although difficult, is essential for proper clinical management including syndrome specific management where appropriate. This work aimed to investigate the molecular basis of disease in a single proband born to consanguineous parents, who presented with microphthalmia, persistent foetal vasculature, posterior lens vacuoles, vitreoretinal dysplasia, microcephaly, hypotelorism and global developmental delay, and was registered severely visually impaired by 5 months of age.
Methods: Extensive genomic pre-screening, including microarray comparative genomic hybridisation and sequencing of a 114 gene panel associated with cataract and congenital ophthalmic disorders was conducted by an accredited clinical laboratory. Whole exome sequencing (WES) was undertaken on a research basis and in vitro TOPflash transcriptional reporter assay was utilised to assess the impact of the putative causal variant.
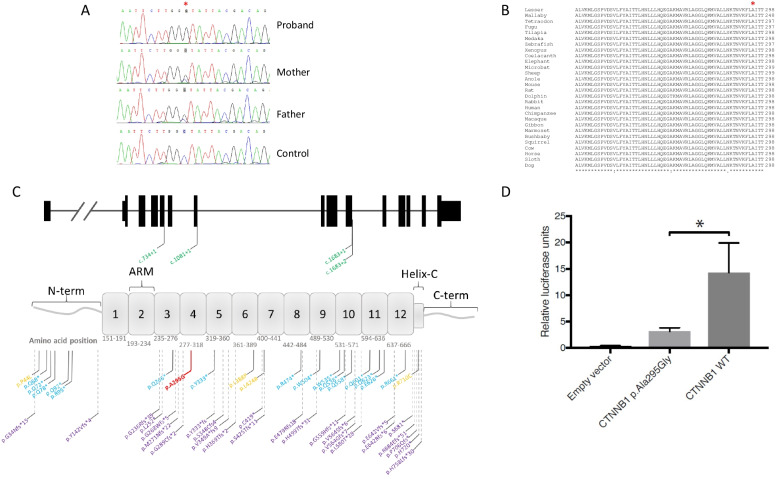
Results: In the proband, WES revealed a novel, likely pathogenic homozygous mutation in the cadherin-associated protein beta-1 gene (CTNNB1), c.884C>G; p.(Ala295Gly), which encodes a co-effector molecule of the Wnt/β-catenin pathway. The proband's parents were shown to be heterozygous carriers but ophthalmic examination did not detect any abnormalities. Functional assessment of the missense variant demonstrated significant reduction of β-catenin activity.
Conclusions: This is the first report of a biallelic disease-causing variation in CTNNB1. We conclude that this biallelic, transcriptional inactivating mutation of CTNNB1 causes a severe, syndromic form of microphthalmia, persistent foetal vasculature and vitreoretinal dysplasia that results in serious visual loss in infancy.
Keywords: Beta-catenin; CTNNB1; Developmental delay; Microphthalmia; Recessive; Syndromic; Vitreoretinal dysplasia.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Kuechler A, Willemsen MH, Albrecht B, Bacino CA, Bartholomew DW, van Bokhoven H, et al. De novo mutations in beta-catenin (CTNNB1) appear to be a frequent cause of intellectual disability: expanding the mutational and clinical spectrum. Hum Genet. 2015;134(1):97–109. doi: 10.1007/s00439-014-1498-1. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
