

Whole-genome sequencing of 1,171 elderly admixed individuals from São Paulo, Brazil
- PMID: 35246524
- PMCID: PMC8897431
- DOI: 10.1038/s41467-022-28648-3
Whole-genome sequencing of 1,171 elderly admixed individuals from São Paulo, Brazil
Erratum in
-
Author Correction: Whole-genome sequencing of 1,171 elderly admixed individuals from Brazil.Nat Commun. 2022 Mar 30;13(1):1831. doi: 10.1038/s41467-022-29575-z. Nat Commun. 2022. PMID: 35354829 Free PMC article. No abstract available.
Abstract
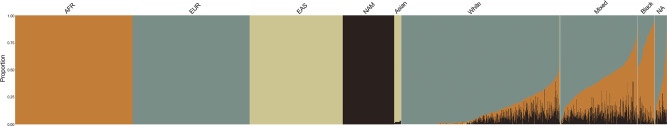
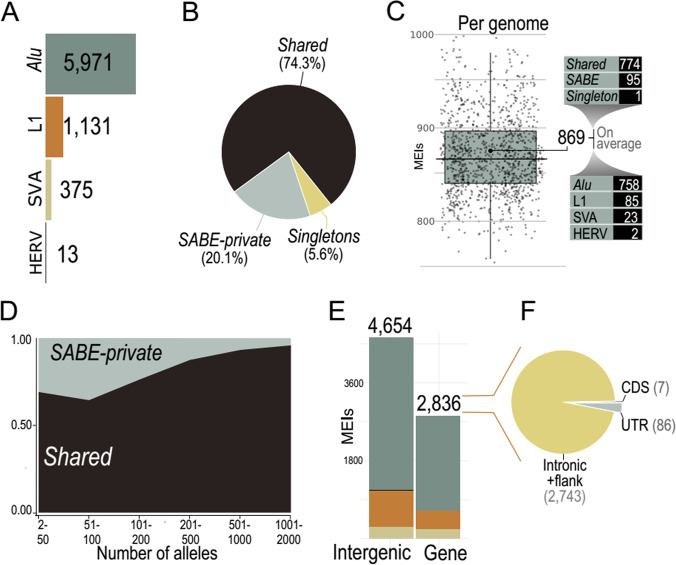
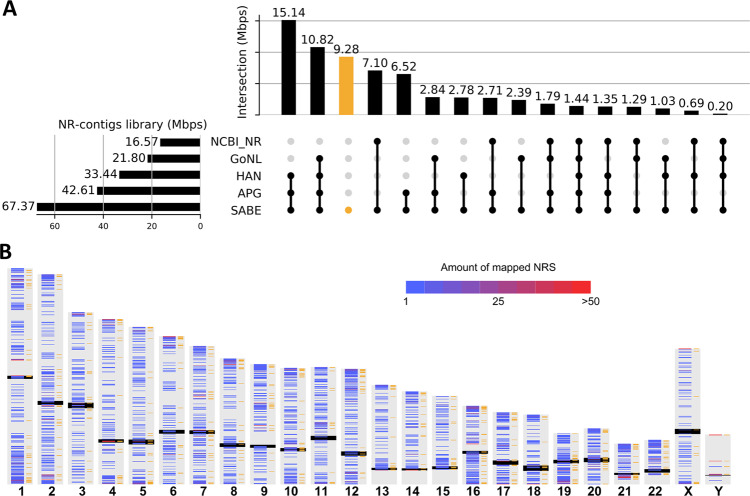
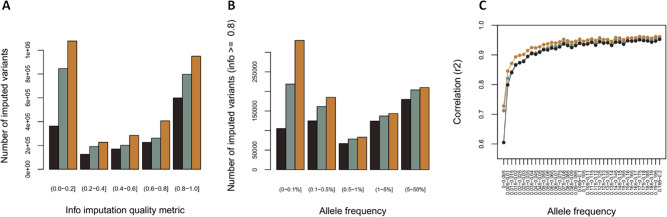
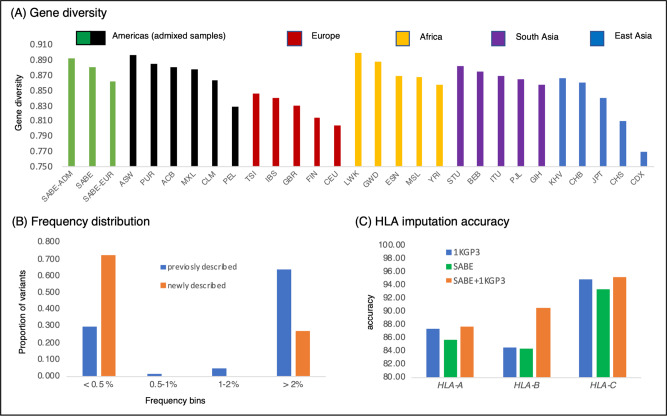
As whole-genome sequencing (WGS) becomes the gold standard tool for studying population genomics and medical applications, data on diverse non-European and admixed individuals are still scarce. Here, we present a high-coverage WGS dataset of 1,171 highly admixed elderly Brazilians from a census-based cohort, providing over 76 million variants, of which ~2 million are absent from large public databases. WGS enables identification of ~2,000 previously undescribed mobile element insertions without previous description, nearly 5 Mb of genomic segments absent from the human genome reference, and over 140 alleles from HLA genes absent from public resources. We reclassify and curate pathogenicity assertions for nearly four hundred variants in genes associated with dominantly-inherited Mendelian disorders and calculate the incidence for selected recessive disorders, demonstrating the clinical usefulness of the present study. Finally, we observe that whole-genome and HLA imputation could be significantly improved compared to available datasets since rare variation represents the largest proportion of input from WGS. These results demonstrate that even smaller sample sizes of underrepresented populations bring relevant data for genomic studies, especially when exploring analyses allowed only by WGS.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Taliun, D. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. bioRxiv. https://www.biorxiv.org/content/10.1101/563866v1 (2019). - DOI - PMC - PubMed
-
- Li J, et al. Decoding the genomics of abdominal aortic aneurysm. Cell. 2018;174:1361–1372 e10. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
