

Osteopontin aggravates acute lung injury in influenza virus infection by promoting macrophages necroptosis
- PMID: 35246529
- PMCID: PMC8897470
- DOI: 10.1038/s41420-022-00904-x
Osteopontin aggravates acute lung injury in influenza virus infection by promoting macrophages necroptosis
Abstract
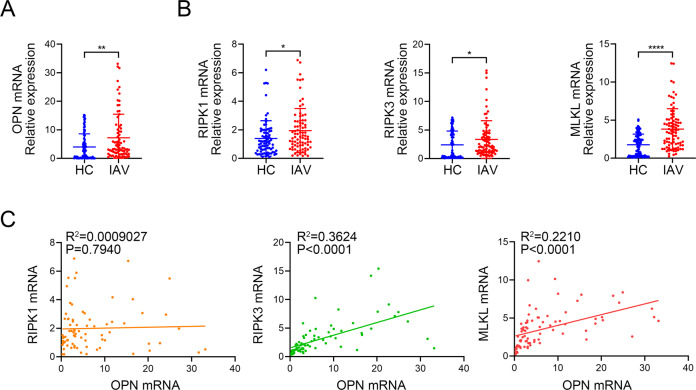
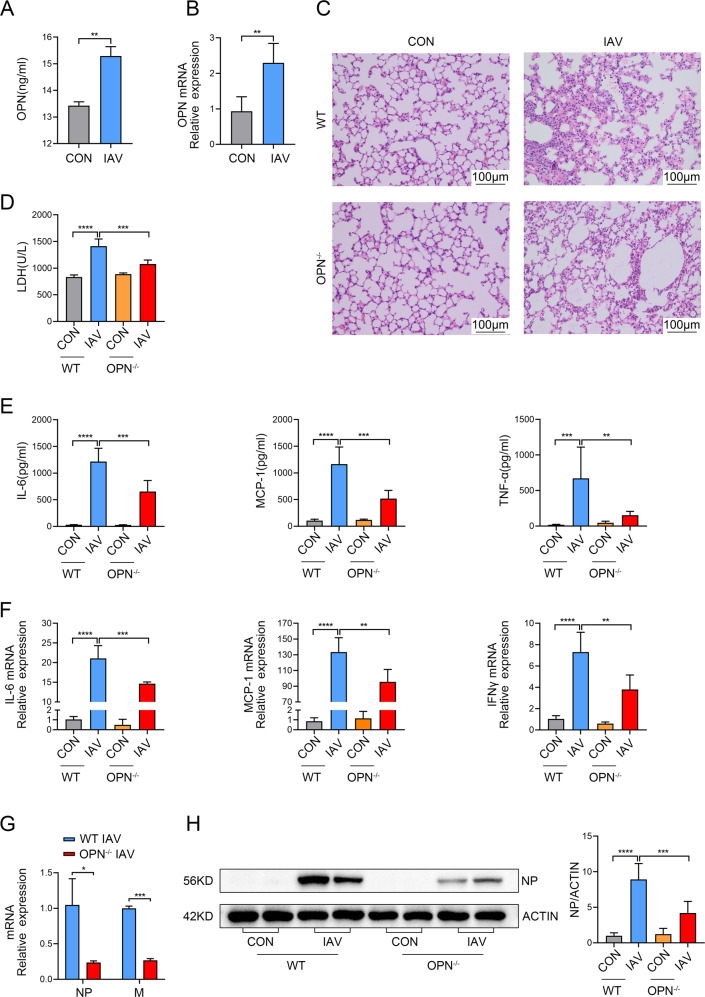
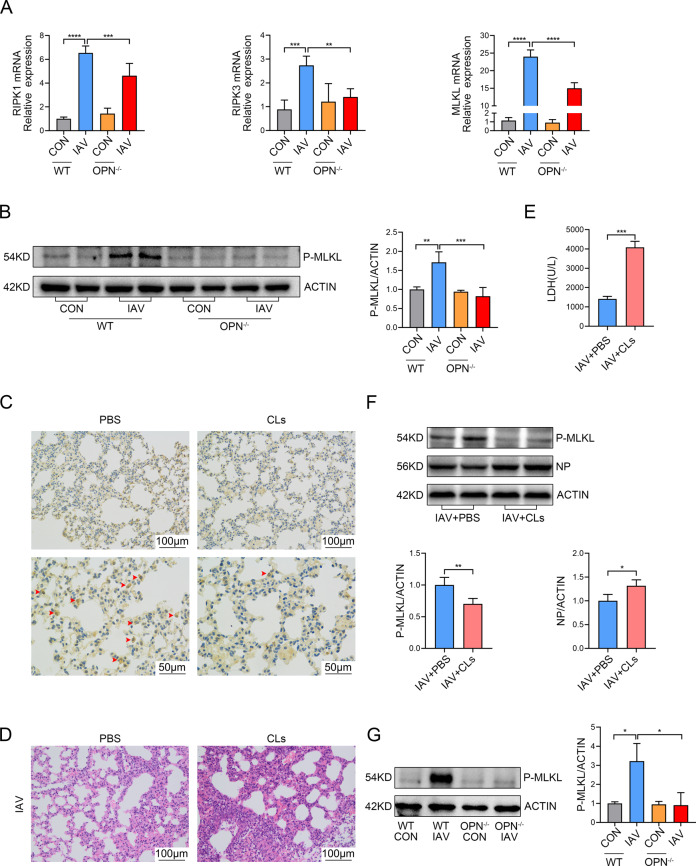
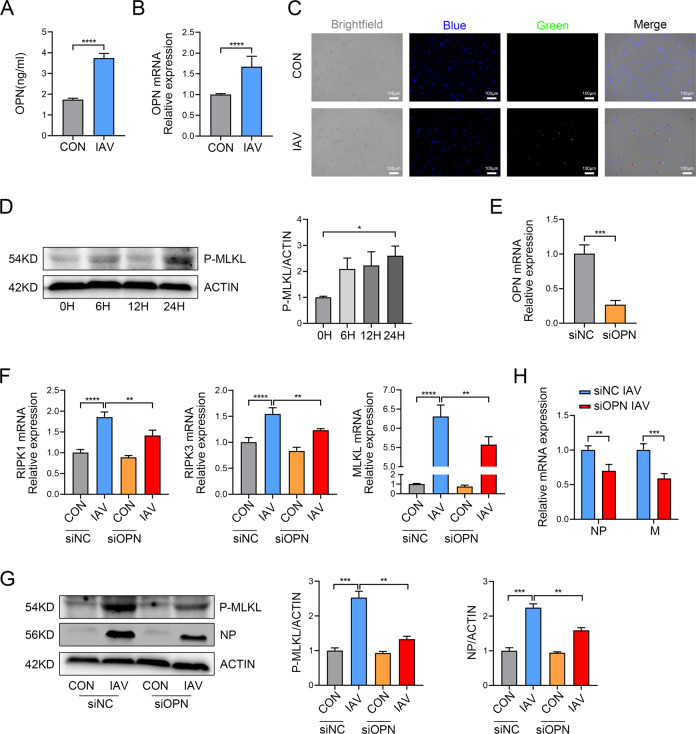
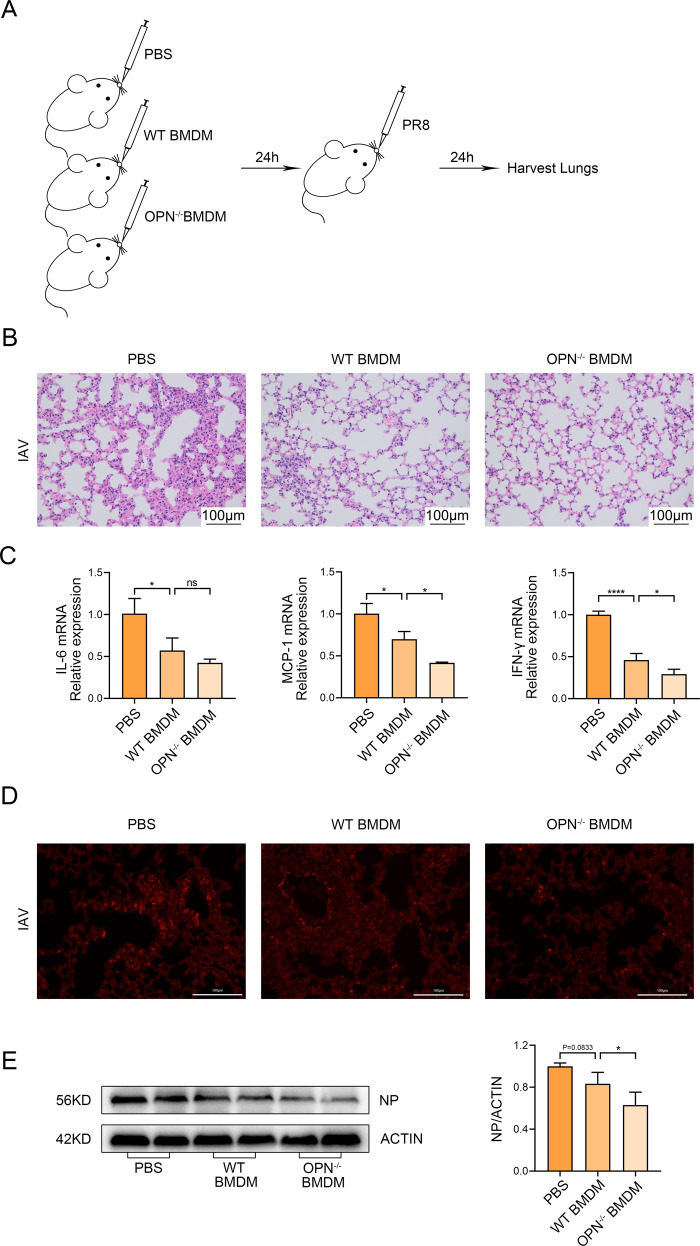
Infection with influenza A virus (IAV) can trigger pulmonary inflammation and lung damage. Osteopontin (OPN) is an essential regulator of cell death and immunity. However, the role and underlying mechanism of OPN in cell death in IAV-induced pulmonary injury remain poorly understood. Here, we demonstrated that OPN-deficient (OPN-/-) mice were insensitive to IAV, exhibiting decreased viral loads and attenuated lung injury after IAV infection compared to those in wild-type (WT) mice. Moreover, macrophage necroptosis was significantly reduced in OPN-/- mice infected with IAV compared to that in infected WT mice. OPN increased the expression of necroptosis-related genes and exacerbated macrophage necroptosis in IAV-infected THP1 cells. Notably, adoptive transfer of WT bone marrow-derived macrophages (BMDMs) or OPN-/- BMDMs into mice restored resistance to influenza infection, and the rescue effect of OPN-/- BMDMs was better than that of WT BMDMs. Collectively, these results suggest that OPN deficiency in macrophages reduces necroptosis, which leads to a decrease in viral titers and protects against IAV infection. Therefore, OPN is a potential target for the treatment of IAV infection.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
LinkOut - more resources
Full Text Sources
Research Materials
